Neuroimaging in Frontotemporal Dementia & Clinical Trials
The utility of MRI & PET imaging biomarkers in our understanding of Frontotemporal Dementia (FTD) variants, and their use as endpoints in FTD clinical trials.
What are the complexities of clinical diagnosis and proteinopathy (Tau vs. TDP-43) in FTD?
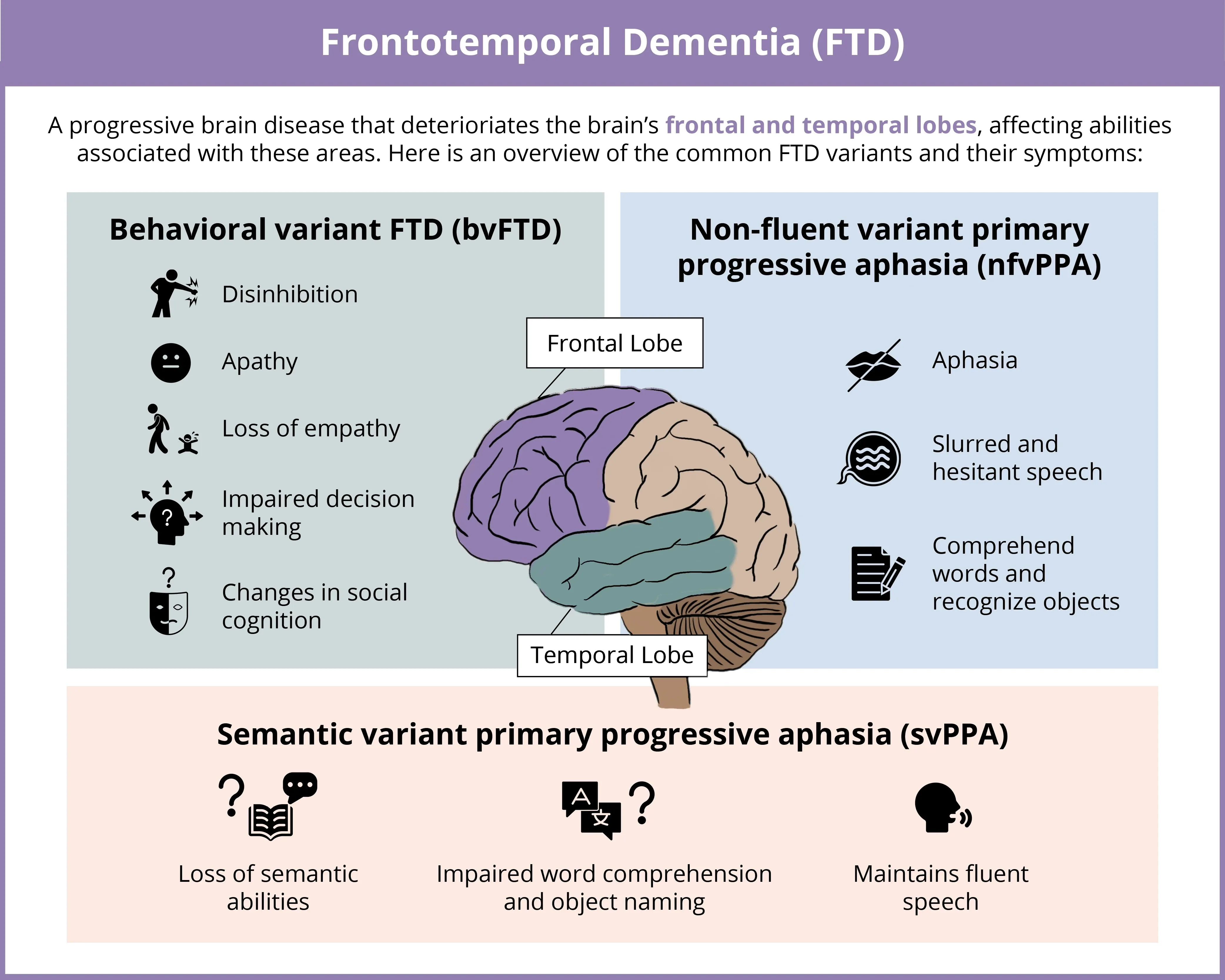
FTD Subtypes
Frontotemporal dementia (FTD) is a progressive, heterogeneous (clinical and pathologic) group of neurodegenerative disorders characterized by degeneration of the frontal and temporal lobes of the brain. The three main clinical subtypes are behavioral variant FTD (bvFTD), non-fluent variant primary progressive aphasia (nfvPPA), and semantic variant primary progressive aphasia (svPPA). bvFTD is the most prevalent FTD subtype, accounting for approximately 70% of cases (Hogan, 2016; Leroy, 2021). The main clinical symptoms of bvFTD include behavioral changes, such as disinhibition, apathy, and impaired social cognition (Rascovsky, 2011). In contrast, nfvPPA and svPPA are less prevalent, with nfvPPA accounting for roughly 25% of cases. nfvPPA and svPPA differentially impair speech and language processes (Gorno-Tempini, 2011). Specifically, nfvPPA is linked to incorrect use of grammar or syntax and speech deficits, with speech progressively becoming nonfluent, and may result in mutism. In contrast, svPPA is characterized by impaired semantic comprehension, specifically for single words, yet speech production is generally not impaired (Gorno-Tempini, 2011).
 Click to copy link
Click to copy link
Complexities of Clinical Diagnosis
FTD is the second leading cause of early-onset dementia after Alzheimer’s disease (AD), representing roughly 10% of dementia cases in those younger than 65 years of age (Hogan, 2016). However, these estimates may be inaccurate since FTD is often misdiagnosed or diagnosed later than it should be. This ambiguity is largely due to the diagnosis of FTD depending on detection of clinical features. The heterogeneity of FTD clinical symptoms and their overlap with those of various psychiatric disorders makes FTD difficult to diagnose. In particular, the clinical symptoms of bvFTD, including apathy, can easily be misinterpreted as depression, putting bvFTD patients at an increased likelihood of misdiagnosis (Tsoukra, 2022). Furthermore, the genetic and molecular pathologies associated with FTD overlap with those of other neurological disorders, such as progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), and amyotrophic lateral sclerosis (ALS). Therefore, identifying disease-specific biomarkers, such as neuroimaging markers, is crucial for achieving earlier and more accurate diagnosis. For instance, MRI atrophy biomarkers have shown significant effect sizes and account for a considerable percentage of variance in clinical function, supporting their use as outcome measures and for patient classification in clinical trials (Staffaroni, 2019). In addition, recent advances in PET imaging with specific radioligands, including those binding to tau and TDP-43, will facilitate the development of disease-modifying therapies for FTD, which are currently unavailable, and allow for earlier interventions when they may be most effective.
Neuropathologic Features
The fundamental pathologic process underlying FTD is frontotemporal lobar degeneration (FTLD), characterized by neurodegeneration and associated brain atrophy, as well as gliosis in the frontal and temporal lobes (Hernandez, 2018). FTLD can be classified into three main subtypes identified post-mortem: FTLD-tau, FTLD-TDP, and FTLD-FUS. These subtypes are distinguished by the specific aggregation of misfolded proteins in neurons and glial cells, primarily involving tau, the transactive response DNA binding protein of 43 kDa (TDP-43), and fused in sarcoma (FUS) protein (Liu, 2019).
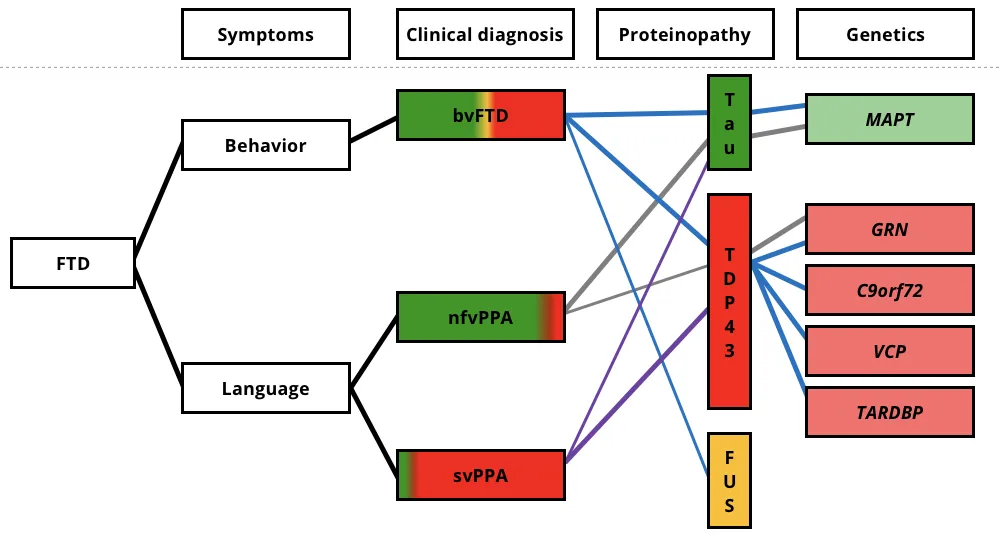
The clinical, pathological, and genetic associations for the three FTD variants. Clinical symptoms are shown on the left, with the colored regions of clinical diagnosis representing the relative percentages of patients found to have each underlying neuropathologic proteinopathy. The colored lines connect the associated clinical, pathologic, and genetic features. bvFTD, behavioral-variant frontotemporal dementia; svPPA, semantic-variant primary progressive aphasia; nfvPPA, non-fluent variant primary progressive aphasia; FUS, fused in sarcoma; TDP-43, transactive response (TAR) DNA-binding protein of 43 kDa; C9orf72, chromosome 9 open reading frame 72; GRN, progranulin mutations; MAPT, microtubule-associated protein tau; and VCP, valosin-containing protein. Figure reproduced from Liu et al. (Liu, 2019) under the Creative Commons Attribution License.
Interestingly, there is not a direct correlation between FTLD subtypes and clinical diagnosis. For example, approximately 50% of bvFTD cases show intracellular TDP-43 inclusions, while roughly 40% are associated with tau pathology (Chare, 2014; Mann, 2017). While most cases of FTD are sporadic, about 30% have a familial history of dementia. Among inherited cases, around 60% exhibit specific proteinopathies, such as MAPT mutations linked to FTLD-tau and C9orf72, GRN, VCP, and TARDBP mutations associated with FTLD-TDP (Le Ber, 2013).
FTLD-tau accounts for about 40% of FTLD cases and is characterized by the abnormal aggregation of tau protein in neuronal and/or glial cells. Tau pathology in genetic forms of FTD linked to MAPT mutations is heterogeneous, with most cases exhibiting 4-repeat (4R) tau pathology, and some cases forming neurofibrillary tangles similar to those found in Alzheimer’s disease (AD) (Levy, 2022). Tauopathies are also associated with various other neurodegenerative disorders, including CBD and PSP. FTLD-tau is commonly observed in approximately 40% of bvFTD cases and most nfvPPA cases, but it is rare in svPPA.
FTLD-TDP constitutes roughly 50% of FTLD cases, making it the most common subtype. TDP-43, a nuclear protein involved in exon skipping and transcription regulation, becomes ubiquitinated and aggregates to the cytoplasm in disease states, forming cytoplasmic inclusions (Neumann, 2006). TDP-43 pathology is also found in most patients with ALS (Neumann, 2013). FTLD-TDP has four primary pathological subtypes more closely linked to their genetic forms than to clinical phenotypes: Type A is associated with GRN gene mutations and linked to bvFTD and nfvPPA; Type B is associated with C9orf72 mutations and linked to bvFTD and ALS; Type C is linked to most cases of svPPA; and Type D is associated with VCP gene mutations and linked to bvFTD and ALS (Neumann, 2021). Recent advances in TDP-43 PET imaging tracers show promise for enhancing diagnosis and treatment of FTLD-TDP (Cordts, 2023; Seredenina, 2023).
FTLD-FUS is the rarest subtype, representing about 5-10% of FTLD cases. It is characterized by the deposition of FUS protein and can present as sporadic, early-onset bvFTD, marked by severe and rapidly progressive neuropsychiatric symptoms. Mutations in the FUS gene, which encodes the FUS protein, are also implicated in ALS (Vance, 2009).
Which imaging biomarkers can be used to distinguish subtypes of FTD and/or distinguish FTD from AD?
The specificity and sensitivity of diagnosing FTD can be significantly improved using imaging biomarkers, such as magnetic resonance imaging (MRI) and positron emission tomography (PET). In addition to these techniques, MRI modalities such as diffusion tensor imaging (DTI) and arterial spin labeling (ASL), are effective for diagnosing FTD by assessing white matter integrity and quantifying cerebral perfusion, respectively; however, the specificity and sensitivity of these techniques can vary (Zhang, 2013; Ono, 2016; Tosun, 2016; Watanabe, 2021). These imaging methods are valuable tools for identifying brain changes associated with FTD, thereby aiding in earlier and more accurate diagnosis. Longitudinal studies of brain changes can also provide meaningful outcome measures for clinical trials. Differentiating between FTD subtypes and distinguishing FTD from other dementias, such as AD, is essential for accurate diagnosis, prognosis, and treatment, as well as for proper classification in clinical trials.
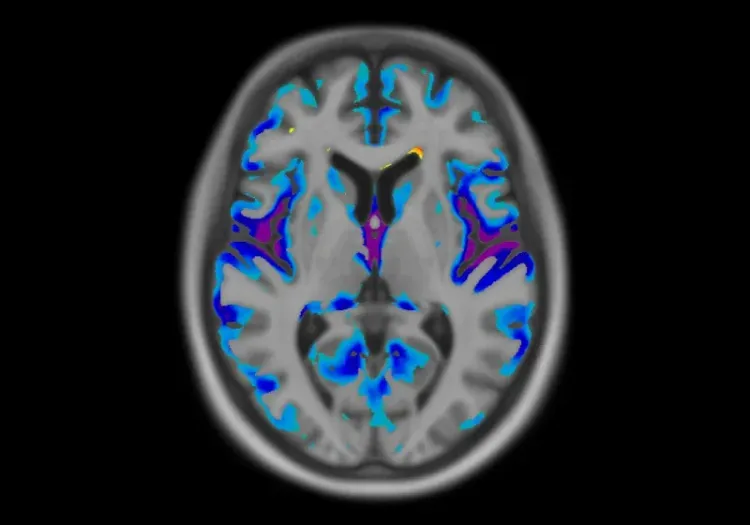
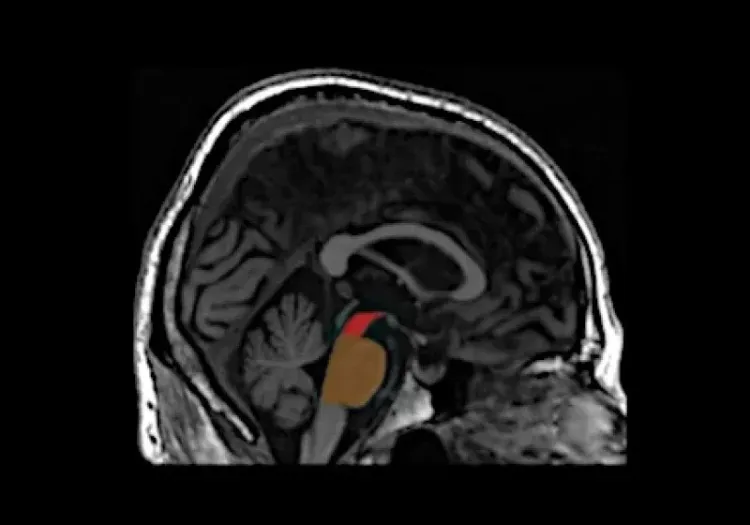
MRI is an important tool for investigating atrophy patterns associated with FTD, allowing for differentiation from other dementias, such as AD. Typically, FTD presents with atrophy in the anterior temporal and frontal lobes, while AD is characterized by generalized atrophy, particularly in the medial temporal lobe (Bocti, 2006; Chouliaras, 2023). An anterior-posterior atrophy gradient in the temporal lobe is indicative of FTLD rather than AD (Harper, 2014). Additionally, bvFTD is associated with reduced gray matter volume in the caudate, which effectively distinguishes it from AD with an accuracy of 79% (Frings, 2014). A study exploring the relationship between white matter hyperintensities (WMH) and gray matter atrophy found that FTD is more closely linked to changes in frontal regions and basal ganglia, unlike the insular and parieto-occipital regions affecting AD (Dadar, 2021). Longitudinal MRI studies have shown that whole brain and ventricular volumes can be reliably tracked over one year, correlating changes with declines in clinical measures (Knopman, 2009). Moreover, the annual decline in gray matter volume in the lateral orbitofrontal gyrus and in temporal white matter is more pronounced in bvFTD compared to AD, highlighting differing atrophy rates (Frings, 2014). Together, these findings suggest that MRI atrophy patterns and longitudinal volumetric assessments can effectively differentiate FTD from AD, linking FTD to reductions in the anterior temporal and frontal lobes, as well as subcortical structures, such as the basal ganglia.
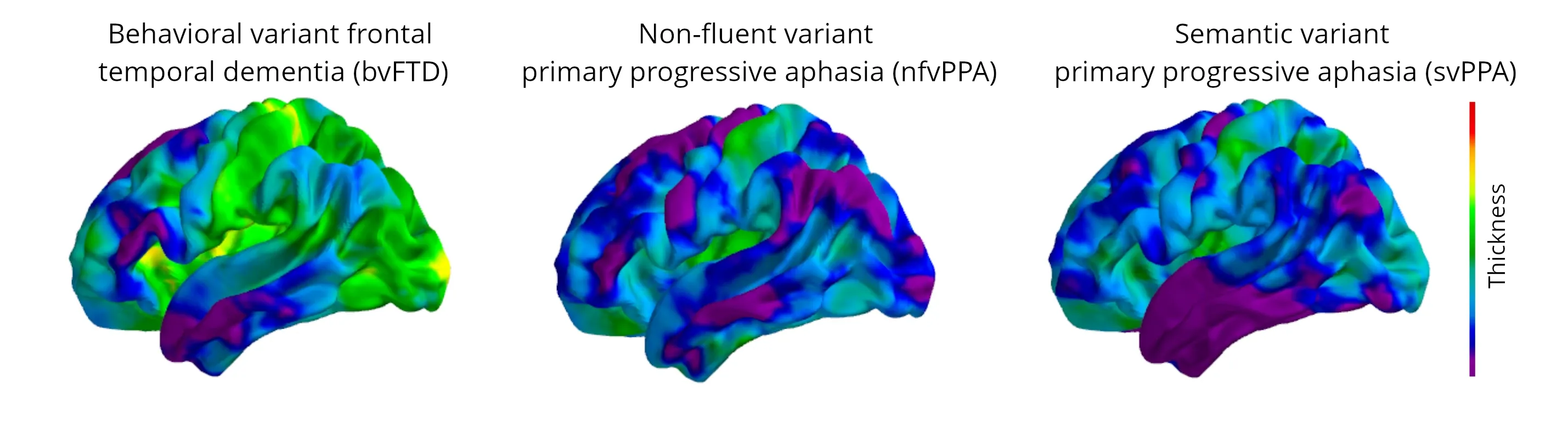
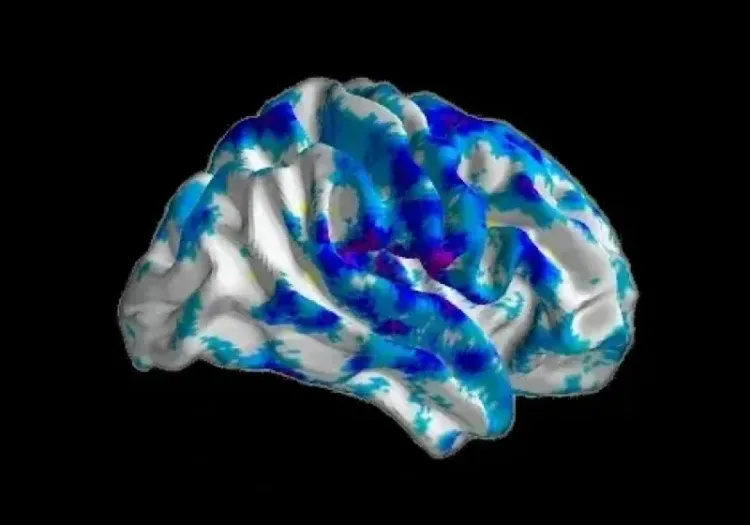
Progressive changes in cortical thickness over a 12-month period are illustrated for each variant, demonstrating the differential regional changes, with the greatest atrophy in the purple and blue colors (data generated by Biospective).
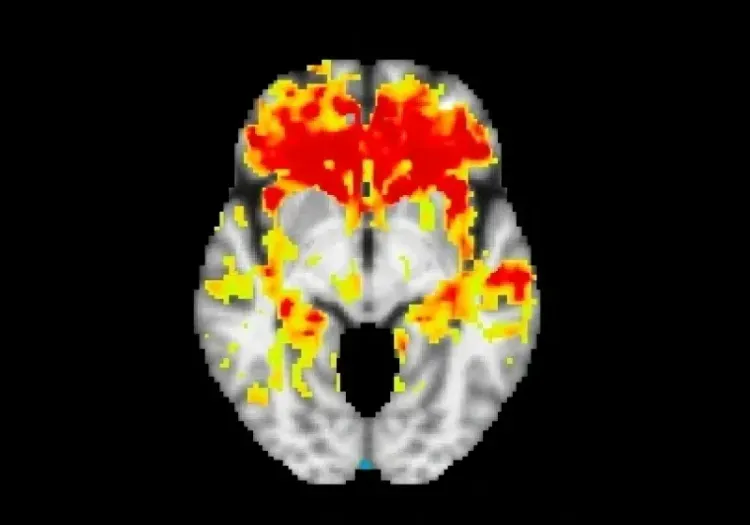
[18F]2-fluoro2-deoxyglucose (FDG) PET imaging is particularly effective in diagnosing FTD, demonstrating high sensitivity and specificity. A comparison study found that SPECT/PET scans have greater diagnostic accuracy for FTD than MRI (Mendez, 2007). FDG PET patterns clearly distinguish FTD from AD, showing diffuse posterior temporal-parietal hypometabolism in AD and focal frontal and anterior temporal hypometabolism in FTD (Mosconi, 2008; Gordon, 2016). The use of amyloid PET tracers further enhances the differentiation between FTD and AD, reinforcing the role of amyloid-β pathology in AD while indicating its absence in FTD (Hellwig, 2019). Conversely, tau pathology is present in both AD and FTLD-tau, as well as other neurodegenerative diseases (e.g. PSP and CBD). Recent studies using tau PET imaging have shown promising results for FTD patients with MAPT mutations, revealing stronger binding in regions such as the right inferior temporal lobe, left putamen, left medial orbitofrontal lobe, and hippocampus (Levy, 2022). Advancements in PET ligands targeting TDP-43 also show potential for improving diagnostic accuracy and treatment development for FTLD-TDP (Seredenina, 2023).
Distinct atrophy patterns are observed across the different FTD subtypes. For instance, bvFTD typically involves atrophy in the frontal lobe, anterior insula, and anterior cingulate, with or without temporal lobe atrophy. In contrast, nfvPPA is characterized by left-sided perisylvian atrophy, affecting the superior temporal, insular, and frontal operculum, while svPPA usually presents with left-sided anterior temporal atrophy, particularly affecting the temporal pole and hippocampus (Galton, 2001; Antonioni, 2023; Tartaglia, 2023). Both nfvPPA and svPPA exhibit asymmetrical atrophy, with more pronounced changes in the left hemisphere. However, in svPPA, the atrophy of the left temporal lobe progresses more rapidly, whereas in nfvPPA, the left frontal lobe atrophy shows the fastest rate of decline (Rohrer, 2012).
Both FTLD-tau and FTLD-TDP show widespread frontal and anterior temporal gray matter loss, but FTLD-tau is associated with a higher relative white matter disease burden, as confirmed by DTI (McMillan, 2013). Furthermore, FTD patients with GRN mutations exhibit a significantly greater annual rate of whole brain atrophy compared to those with MAPT mutations, suggesting that GRN mutations are linked to more widespread cerebral atrophy, whereas MAPT mutations are associated with localized antero-medial temporal atrophy (Whitwell, 2011). Conversely, C9orf72 mutations are associated with lower rates of volumetric decline compared to both MAPT and GRN mutations, although clinical variability among patients can still be significant (Gordon, 2016).
Our team would be happy to answer any questions about Neuroimaging in Frontotemporal Dementia & Clinical Trials or provide specific information about our other Imaging services.
Discover more about our Imaging Services
Related Content
Up-to-date information on best practices related to the use of neuroimaging in clinical trials of neurological diseases.
Modeling Protein Misfolding in Neurological Diseases
Computational modeling of protein misfolding mechanisms in neurodegeneration and the application to discovery & development of disease-modifying therapeutics.
Diffusion MRI & Frontotemporal Dementia (FTD)
Diffusion neuroimaging analysis from the FTLDNI natural history study of Frontotemporal Dementia (FTD).
MRI & Corticobasal Degeneration (CBD)
Longitudinal MRI brain atrophy measures from the 4RTNI and FTLDNI studies including sample size calculations for clinical trials of Corticobasal Degeneration.
Imaging Biomarkers to Distinguish CBD from Other Tauopathies
Overview of brain imaging biomarkers to identify Corticobasal Degeneration (CBD) subjects and their use in clinical trials of disease-modifying therapeutics.
Imaging Biomarkers for Progressive Supranuclear Palsy (PSP)
An overview of the various brain imaging methods (MRI, PET, SPECT) available to assess efficacy of disease-modifying therapeutics in clinical trials of PSP.
MRI Measures of Disease Progression for PSP Clinical Trials
MRI biomarkers (including brain atrophy) from the 4RTNI and FTLDNI natural history studies of Progressive Supranuclear Palsy (PSP).
Longitudinal Change in Tau PET in MCI & Alzheimer’s Disease
An overview of the natural history of change in Tau PET tracer uptake & binding in Mild Cognitive Impairment (MCI) & Alzheimer’s disease (AD).
MRI in Clinical Trials of Multiple System Atrophy (MSA)
This resource provides an overview of the utility of volumetric MRI and diffusion-weighted imaging (DWI) as biomarkers in research studies of MSA.
Frontotemporal Dementia (FTD) & MRI Brain Atrophy
Neuroimaging biomarkers (including MRI brain atrophy) from the FTLDNI natural history study of Frontotemporal Dementia (FTD).