Modélisation du mauvais repliement des protéines dans les maladies neurologiques
Modélisation informatique des mécanismes de mauvais repliement des protéines dans la neurodégénérescence et application à la découverte et au développement de traitements modificateurs de la maladie.
Quel rôle jouent les protéines mal repliées dans les maladies neurologiques?
Introduction à la dynamique des protéines mal repliées
Les protéines sont de grosses molécules composées d'acides aminés et leur configuration de repliement correcte assure une stabilité à long terme aux environnements biologiques. En revanche, un défaut de repliement correct est généralement associé à une variété de conditions pathologiques (Dobson, 2003; Nassar, 2021). Par conséquent, les protéines qui ne se configurent pas correctement sont appelées protéines mal repliées (MP) et sont considérées comme le processus initial qui déclenche de nombreux troubles, tels que la maladie d'Alzheimer, la maladie de Parkinson, la sclérose latérale amyotrophique (SLA), la maladie de Creutzfeldt-Jakob (MCJ) et plusieurs autres troubles neurodégénératifs humains (Scheckel, 2018; Mehra, 2019; Lövestam, 2023; Michaels, 2023).
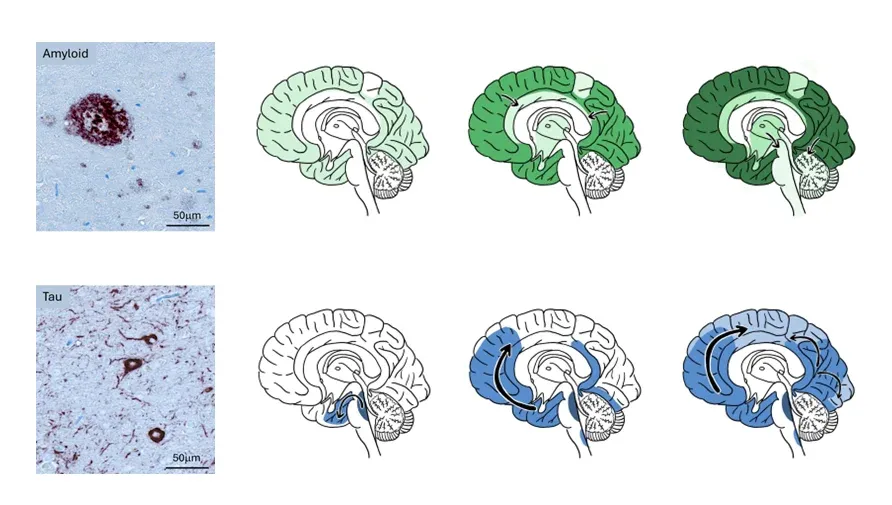
La maladie d'Alzheimer se caractérise par différents modèles microscopiques de protéines mal repliées Aß et tau qui produisent des modèles anatomiques caractéristiques de progression des agrégats Aß et tau dans le cortex.
Il existe une série de mécanismes intrinsèques entourant le processus de mauvais repliement des protéines qui doivent être entièrement caractérisés afin de comprendre pleinement leur effet toxique dans les cellules (Eisele, 2016; Lewis, 2016). Ces mécanismes comprennent l'ensemencement, l'agrégation, la propagation et/ou l'étalement des MP. Ainsi, les signatures spécifiques de l'évolution temporelle de ces mécanismes liés aux MP sont généralement appelées « dynamique des MP ». Le principal obstacle à l'identification d'événements déclencheurs spécifiques aux maladies conformationnelles est la capacité d'extrapoler l'impact de la dynamique des MP au niveau moléculaire à des échelles plus macroscopiques.
Modèles mathématiques pour les protéines mal repliées
Les modèles mathématiques sont des environnements théoriques qui visent à simuler et à reproduire autant que possible la dynamique réelle des processus biologiques. Les modèles mathématiques exprimés par des équations différentielles sont un choix naturel car ils peuvent facilement reproduire la dynamique non linéaire et complexe de plusieurs processus interagissant dans le temps. Dans le contexte des protéines mal repliées, les modèles mathématiques peuvent être classés en deux grandes catégories. Une classe de modèles visant à simuler des processus au niveau moléculaire tels que l'ensemencement, l'agrégation et la propagation spatiale de cellule à cellule à courte distance, et une classe de modèles qui simulent des interactions plus macroscopiques telles que la propagation de MP à longue distance dans le cerveau humain. Cette catégorisation permet de constater que les modèles à l'échelle microscopique ont été principalement développés dans le domaine de la cinétique chimique. En revanche, les modèles à plus grande échelle (c'est-à-dire macroscopiques) ont surtout été proposés dans le cadre d'études de neuro-imagerie.
Dynamique de type prion
La maladie du prion pouvant également être considérée comme un processus MP (Scheckel, 2018), il était naturel que les premiers modèles mathématiques pour la MP simulent les mécanismes biologiques de réplication et d'agrégation du prion (Eigen, 1996). Basé sur des équations différentielles ordinaires (ODE), le modèle prototype initial qui a été largement accepté est actuellement connu sous le nom de modèle de polymérisation nucléée (NPM) (Nowak, 1998) et s'est concentré sur les mécanismes d'autocatalyse qui visaient à expliquer la conversion d'une protéine prion typique en une protéine d'agent infectieux. Il est remarquable que Nowak et al. (Nowak, 1998) se soient rendu compte que les mécanismes de réplication et d'agrégation des protéines inclus dans le NPM étaient analogues à la dynamique que l'on trouve dans les modèles épidémiologiques les plus courants pour décrire la dynamique des virus. Il n'est donc pas surprenant que les modèles macroscopiques modernes décrivant la propagation des MP aient également été inspirés par des modèles épidémiologiques bien fondés.
En établissant le NPM pour la dynamique des prions comme pierre angulaire, plusieurs études (Petkova, 2005; Frost, 2009) ont démontré que les agrégats de fibrilles des protéines α-synucléine, tau et Aß s'autopropagent selon des mécanismes biochimiques analogues à ceux décrits pour l'agrégation/propagation des prions. Ces observations, associées à plusieurs modèles animaux in vivo, ont permis de fonder l'hypothèse de la progression des maladies neurodégénératives selon le modèle du prion (Frost, 2010). Selon cette hypothèse, l"infectivité" de la MP se propage à partir de régions initiales contenant une concentration relativement élevée de protéines pathogènes vers d'autres régions cérébrales "non infectées."
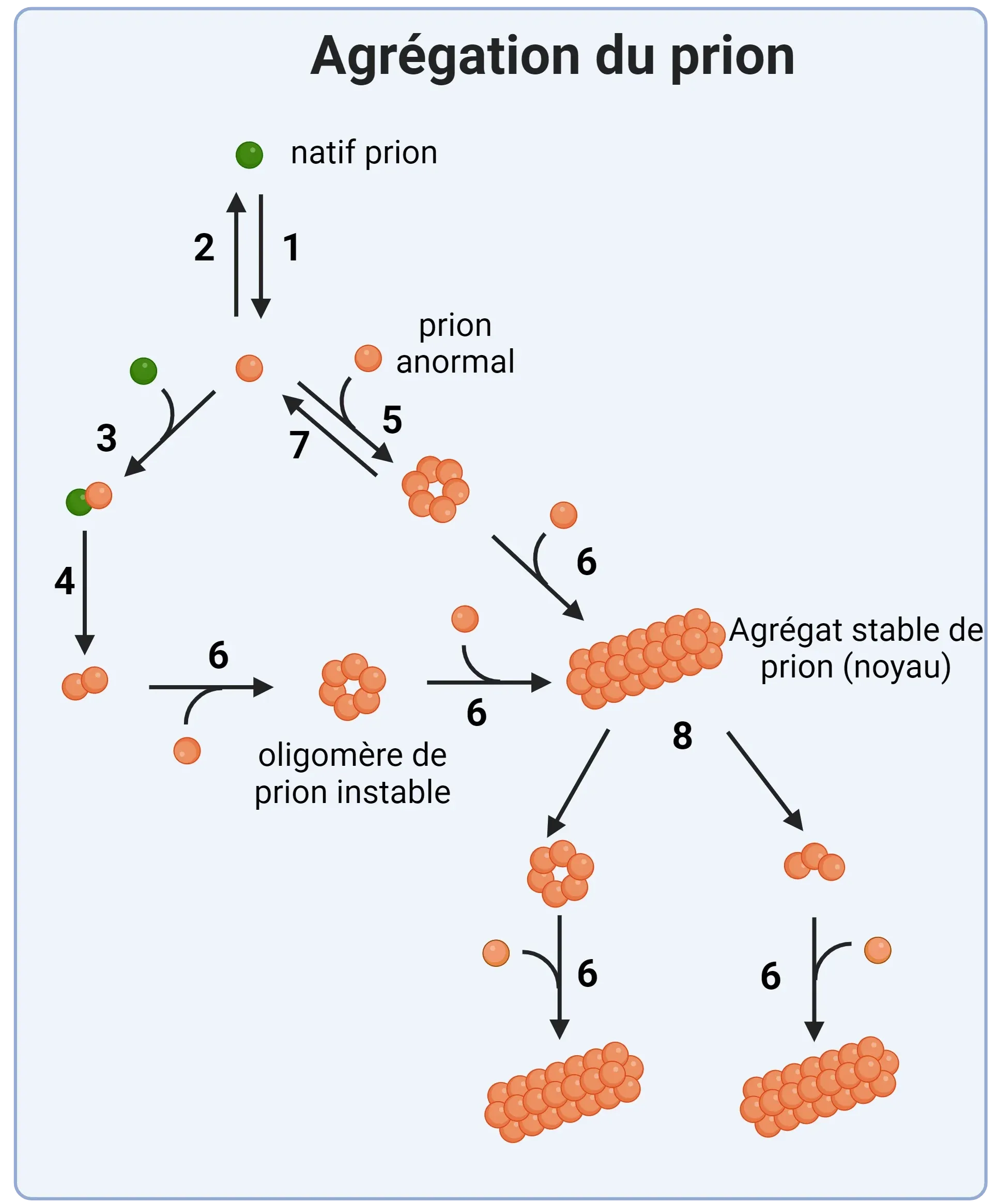
Représentation schématique des mécanismes d'agrégation des prions. Les molécules de prion natives subissent des changements de conformation qui conduisent à une configuration anormale (étape 1). À partir de là, plusieurs étapes d'ensemencement et d'agrégation montrent la formation d'oligomères de prions instables pour finalement produire des agrégats antérieurs stables.
Propagation MP: Propagation spatiale
Payne et al. (Payne, 1998) ont réalisé une percée dans l'étude de la dynamique des PM en fournissant une explication plausible à la latence entre l'apparition de la protéine de l'agent infectieux dans le système nerveux périphérique (SNP) et son apparition dans le système nerveux central (SNC), qui avait été observée précédemment dans des modèles animaux (Scott, 1992). Payne et al. (Payne, 1998) ont donc proposé l'hypothèse que cette latence observée était plus probablement liée à la vitesse de propagation entre des localités voisines, plutôt qu'expliquée par la vitesse à laquelle une protéine prion typique est convertie en protéine d'agent infectieux à un endroit spatial spécifique. Pour prouver une telle hypothèse, la configuration originale de type épidémiologique du NPM a dû être étendue pour intégrer des caractéristiques de « connectivité » spatiale dans l'évolution temporelle de la cinétique des prions (Stumpf, 2000). La principale hypothèse sous-jacente à ce type de modèle étendu était que la protéine de l'agent infectieux se propage dans l'espace le long des axones et des dendrites par un lent transport axonal. En outre, il était naturel de supposer que la vitesse de propagation d'une cellule à l'autre dépendait des forces de connectivité.
Comment la dynamique des protéines mal repliées est-elle modélisée mathématiquement?
La cinétique chimique pour modéliser les mécanismes d'agrégation des protéines
L'approche la plus moderne pour modéliser les protéines mal repliées consiste à utiliser les principes de la cinétique chimique pour dériver des équations maîtresses qui rendent explicitement compte des différents processus microscopiques impliqués dans les mécanismes d'agrégation de la protéine (Cohen, 2012; Cohen, 2013). L'objectif est de caractériser la cinétique de l'agrégation des protéines avec des lois de taux intégrées qui pourraient être dérivées de ces équations maîtresses.
En considérant les phénomènes de croissance filamenteuse et en se basant sur les principes de base des processus homogènes de nucléation, de croissance et de dissociation, Oosawa et al. (Oosawa, 1962) ont proposé le premier prototype de modèles d'équations maîtresses pour l'agrégation des protéines. Comme dans le cas du NPM, les équations maîtresses peuvent être décrites analytiquement par un ensemble infini d'EDO. Cependant, dans la pratique, un tel ensemble infini d'EDO donne lieu à un système fermé de seulement deux EDO qui sont communément appelées les équations de moment et qui caractérisent l'évolution temporelle du nombre et des concentrations de masse des agrégats de protéines, qui sont des quantités directement liées aux mesures des expériences in vitro les plus courantes. Les lois de taux intégrées souhaitées peuvent alors être obtenues directement en résolvant les équations des moments (Cohen, 2013). Cependant, la résolution analytique des équations des moments est une tâche compliquée, en particulier pour l'ensemble du temps de réaction.
Une contribution majeure à la théorie des équations maîtresses pour la formation des protéines a ensuite été apportée par Knowles et al. (Knowles, 2009), qui ont généré des solutions analytiques pour la loi de vitesse intégrée qui étaient valables pour l'ensemble du cours du temps de la réaction. Comme dans le cas de la théorie originale d'Oosawa, ces lois de vitesse intégrées explicites peuvent caractériser la dépendance du temps de réaction à deux paramètres qui peuvent être facilement reliés à des variables phénoménologiques observées expérimentalement (Cohen, 2011). D'un point de vue pratique, il est extrêmement important de disposer d'une formule analytique pour les lois de vitesse intégrées, ce qui permettrait d'adapter les modèles aux données provenant de différentes conditions expérimentales. Par exemple, le modèle proposé par Knowles et al. (Knowles, 2009) a été ajusté aux données in vitro correspondant aux peptides Aß40 et Aß42, ce qui a permis de mieux comprendre la formation des agrégats Aß (Meisl, 2014; Meisl, 2016). Au cours des dernières années, l'approche des équations maîtresses et les lois de taux intégrées correspondantes ont fait l'objet de multiples extensions et généralisations. Une remarquable a été fournie par Cohen et al. (Cohen, 2014), qui, en incluant les mécanismes de propagation spatiale dans les équations maîtresses, ont trouvé qu'une voie d'agrégation secondaire gouverne la vitesse de propagation spatiale par diffusion.
Théorie de la coagulation pour l'agrégation
Comme dans le cas de l'approche des équations maîtresses, la théorie de la coagulation décrite par les équations de Smoluchowski couvre également le cas général de l'auto-association entre des assemblées de particules de tailles différentes. Les premières références aux équations de Smoluchowski dans le contexte de l'agrégation des protéines sont apparues dans Murphy et al. (Murphy, 2000) fpour décrire l'allongement axial des fibrilles par agrégation bout à bout de fibrilles plus courtes. De même, en utilisant les équations de Smoluchowski, Craft et al. (Craft, 2005) ont proposé un modèle de polymérisation dans lequel le processus de nucléation semble implicitement incorporé dans le mécanisme d'association de polymères de petite taille (par exemple monomères, micelles, filaments).
Un tournant important dans le domaine de la modélisation des mécanismes d'agrégation et de propagation des protéines est dû à Achdou et al. (Achdou, 2013). Plutôt que de travailler avec des équations de moment, Achdou et al. (Achdou, 2013) ont tronqué l'ensemble infini d'ODE à un nombre suffisamment grand N. De cette façon, l'équation correspondant au nombre de troncature N devrait être capable de décrire l'évolution temporelle du résumé de tous les assemblages de protéines composés de plus de N monomères. En outre, Achdou et al. (Achdou, 2013) ont réalisé que les équations de Smoluchowski fournissent également un cadre simple pour incorporer des processus de propagation spatiale.
Le développement des techniques d'imagerie modernes exige la possibilité de sonder les modèles à des échelles supérieures aux échelles microscopiques. Dans ce but, un modèle à grande échelle a été proposé par Bertsch et al. (Bertsch, 2017), qui a couplé un ensemble d'équations de Smoluchowski tronquées à une équation de transport de type cinétique qui modélise la propagation des dommages neuronaux par une transmission de type prion de neurone à neurone. Une telle modélisation a alors la capacité d'intégrer deux échelles temporelles différentes évoluant sur le même domaine spatial : une échelle temporelle rapide (par exemple minutes, heures) pour les processus microscopiques (par les équations de Smoluchowski) ; et une échelle lente (par exemple mois, années) pour rendre compte de la progression observée de la MA chez l'homme (par l'équation de transport).
Modélisation de la propagation interrégionale par l'approche réseau
La modélisation de la propagation spatiale des protéines mal repliées par un processus de diffusion homogène n'est pas un choix réaliste dans les grands domaines spatiaux, tels que le cerveau entier. Contrairement à une diffusion homogène, l'approche réseau modélise une propagation rapide de l'infection au sein de groupes de neurones fortement connectés, et une propagation à d'autres groupes par le biais de connexions à longue distance (Matthäus, 2006). Ainsi, à l'aide d'un modèle épidémiologique discret Susceptible-Infecté (SI), Matthäus et al. (Matthäus, 2006) ont caractérisé la propagation de l'infection par la protéine prion le long d'un réseau de neurones interconnectés. Plus important encore, ce cadre mathématique basé sur un réseau peut être facilement adapté pour décrire un scénario à plus grande échelle dans lequel les nœuds du réseau représentent des régions distantes couvrant de vastes domaines spatiaux.
Dans cette lignée, Raj et al. (Raj, 2012) ont proposé un modèle macroscopique de diffusion en réseau (NDM), où le nombre d'afférences MP d'une région cérébrale donnée vers n'importe quelle autre région dépend uniquement des facteurs de concentration de la maladie dans les deux régions et de la force de la connexion anatomique entre elles. En considérant un scénario plus réaliste, Iturria-Medina et al. (Iturria-Medina, 2014) ont introduit un modèle de propagation épidémique (ESM) qui tient compte simultanément de la capacité régionale à produire/nettoyer des MP et de l'information topologique du réseau anatomique du cerveau. Appliqué à l'étude de la maladie d'Alzheimer tardive à l'aide de données TEP sur l'amyloïde, ce modèle a permis de reproduire les schémas de dépôt d'Aß au niveau individuel. L'existence de mécanismes pathologiques détaillés et d'hypothèses pour la progression de la maladie d'Alzheimer a motivé l'examen d'une approche de modélisation multifactorielle plus intégrative qui va au-delà de la formation et de la propagation des protéines mal repliées (Edelstein-Keshet, 2002; Luca, 2003).
Comment les modèles théoriques peuvent-ils être utilisés pour les interventions thérapeutiques?
Généralement, les modèles mathématiques d'agrégation des protéines sont utilisés comme substitut aux interventions thérapeutiques in vitro. L'idée générale est de simuler comment une intervention thérapeutique (par exemple des médicaments, des anticorps, des chaperons moléculaires) peut inhiber certains processus d'agrégation microscopique (Arosio, 2014; Nasica-Labouze, 2015; Kulenkampff, 2021; Ghosh, 2023). Les taux cinétiques d'agrégation des protéines peuvent alors être estimés et comparés dans des conditions naturelles et d'inhibition. Il s'agit d'abord de supposer un changement hypothétique (par exemple, par le traitement) sur les taux cinétiques appropriés, puis de substituer ces taux modifiés dans le modèle original pour simuler les états post-traitement.
Le suivi des taux cinétiques en fonction d'une dose hypothétique de médicament peut aider à extrapoler les petites doses de médicament inhérentes aux environnements in vitro à des doses plus proches des conditions in vivo (Masel, 2000). Dans leur étude pionnière, Masel et al. (Masel, 2000) ont utilisé des modèles mathématiques pour simuler l'inhibition de la propagation de l'amyloïde selon trois approches principales : (i) en abaissant la concentration effective de monomères de protéines non pliées ; (ii) en bloquant les extrémités de polymères en croissance ; et (iii) en augmentant le taux de rupture des polymères. Ils ont constaté que les produits thérapeutiques suivant la deuxième stratégie donneraient des résultats prometteurs, tandis que les autres pourraient être inefficaces ou même accélérer le processus de formation de l'amyloïde à de faibles doses de médicaments.
Simulation de l'inhibition de l'agrégation/propagation de l'amyloïde par trois stratégies différentes: (1) abaissement de la concentration effective en monomères, (2) blocage des extrémités croissantes du polymère et (3) augmentation du taux de rupture du polymère.
Autre exemple, dans le contexte de la protéine bêta-amyloïde, Craft et al. (Craft, 2005) ont établi une relation non linéaire entre le rapport de polymérisation et la charge totale en Aß. Étant donné que le rapport de polymérisation dépend de quatre taux cinétiques différents, plusieurs stratégies de traitement pourraient être testées dans ce contexte : (i) la réduction du taux de production de monomères Aß, (ii) l'augmentation de la fragmentation, (iii) l'augmentation du taux de clairance (c'est-à-dire de dégradation), et (iv) la réduction du taux d'élongation. En simulant les scénarios susmentionnés, Craft et al. (Craft, 2005) ont conclu que tout traitement médicamenteux basé sur des amplificateurs de la vitesse de clairance pourrait être plus efficace pour réduire la charge totale d'Aß que ceux basés sur des amplificateurs de la fragmentation des polymères.
Plus récemment, Arosio et al. (Arosio, 2016) ont utilisé l'approche cinétique chimique pour modéliser l'interaction entre les chaperons moléculaires et différentes espèces de protéines. L'idée principale était d'identifier quelles étapes de la réaction microscopique (c'est-à-dire la nucléation primaire, l'élongation, la fragmentation et la nucléation secondaire) étaient perturbées par la liaison des chaperons moléculaires à certaines espèces de protéines. En utilisant l'approche des équations maîtresses, Arosio et al. (Arosio, 2016) ont comparé les constantes de vitesse cinétiques estimées en l'absence et en présence de différentes concentrations de chaperons moléculaires. Dans le cas de la protéine Aß42, cette analyse a révélé que l'action du chaperon moléculaire appelé DNAJB6 inhibe le processus de nucléation primaire. En revanche, la présence d'un autre chaperon moléculaire (appelé proSP-C Brichos) entraîne une réduction du taux de nucléation secondaire.
Autre exemple, Habchi et al. (Habchi, 2016) ont utilisé l'approche cinétique chimique pour développer une stratégie rationnelle de découverte de médicaments et ont rapporté qu'un médicament anticancéreux (appelé bexarotène) perturbe l'étape de nucléation primaire dans l'agrégation de l'Aß42, retarde la formation d'oligomères toxiques et supprime complètement le dépôt de l'Aß42. Ce cadre général donne lieu à une stratégie systématique de découverte de médicaments visant à identifier une variété de petites molécules (Habchi, 2017) et d'anticorps (Aprile, 2017) qui ciblent non seulement le début de l'agrégation, mais aussi l'étape de nucléation secondaire responsable de la prolifération des oligomères Aß42 toxiques.
Les récentes avancées dans le domaine de la cinétique chimique et de l'agrégation des protéines ont sans aucun doute suscité un regain d'intérêt pour la modélisation des interventions thérapeutiques en tant qu'outil précieux pour la découverte de nouveaux agents thérapeutiques potentiels dans les maladies liées à la MP (Linse, 2020; Kulenkampff, 2021; Ghosh, 2023).
Notre équipe se fera un plaisir de répondre à vos questions sur la modélisation du mauvais repliement des protéines dans les maladies neurologiques ou de vous fournir des informations spécifiques sur nos autres services d'imagerie.
Découvrez nos services d'imagerie
Contenu connexe
Informations actualisées sur les meilleures pratiques liées à l'utilisation de la neuroimagerie dans les essais cliniques sur les maladies neurologiques.
Biomarqueurs TEP dans les essais cliniques sur la maladie de Huntington
Un aperçu de l'utilisation des biomarqueurs d'imagerie TEP pour les essais cliniques de la maladie de Huntington (MH).
Évolution longitudinale de la TEP Tau dans la MCI et la maladie d'Alzheimer
Vue d'ensemble de l'histoire naturelle de l'évolution de l'absorption et de la fixation du traceur de la TEP Tau dans les troubles cognitifs légers (MCI) et la maladie d'Alzheimer (AD).
Biomarqueurs d'imagerie pour distinguer la CBD des autres tauopathies
Vue d'ensemble des biomarqueurs d'imagerie cérébrale permettant d'identifier les sujets atteints de dégénérescence corticobasale (CBD) et de leur utilisation dans le cadre d'essais cliniques de traitements modificateurs de la maladie.
IRM et dégénérescence corticobasale (CBD)
Mesures longitudinales de l'atrophie cérébrale par IRM provenant des études 4RTNI et FTLDNI, y compris le calcul de la taille des échantillons pour les essais cliniques sur la dégénérescence corticobasale.
Démence frontotemporale (DFT) et atrophie cérébrale par IRM
Biomarqueurs IRM (y compris l'atrophie cérébrale) issus des études FTLDNI sur l'histoire naturelle de la démence frontotemporale (DFT).
La neuroimagerie dans la démence frontotemporale et les essais cliniques
L'utilité des biomarqueurs d'imagerie IRM et TEP dans notre compréhension des variantes de la démence frontotemporale (DFT) et leur utilisation comme critères d'évaluation dans les essais cliniques sur la DFT.