错误折叠的蛋白质在神经疾病中扮演什么角色?
错误折叠蛋白动力学简介
蛋白质是由氨基酸组成的大分子,其正确的折叠结构为生物环境提供了长期稳定性。相反,折叠错误通常与多种病理状况有关(Dobson,2003 ;Nassar,2021 )。因此 ,未能正确折叠的蛋白质被称为错误折叠的蛋白质(MP),被认为是引发多种疾病的初始过程,例如阿尔茨海默氏病(AD)、帕金森氏病(PD)、肌萎缩性侧索硬化症(ALS)、克雅氏病(CJD)和其他几种人类神经退行性疾病(Scheckel,2018 ;Mehra,2019; Lövestam,2023 ;Michaels,2023 )。
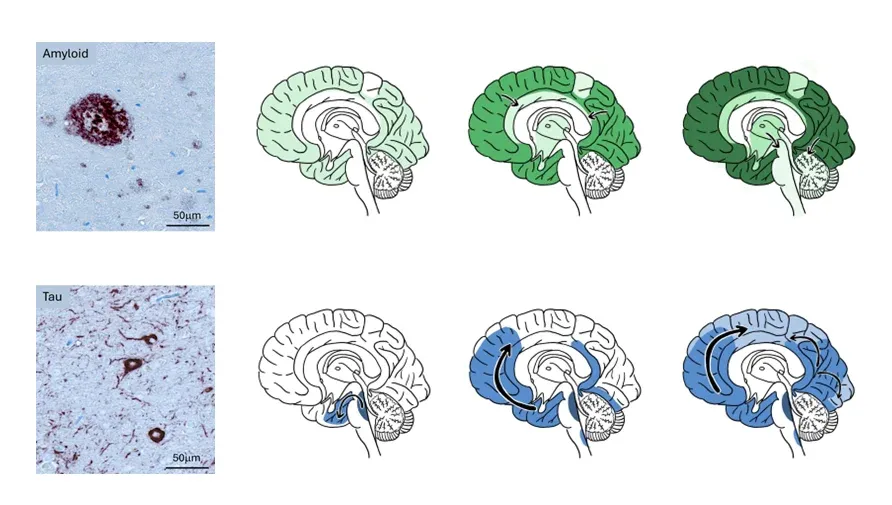
阿尔茨海默氏症的特征是A和Tau蛋白发生错误折叠,形成不同的微观结构,从而在皮层中形成A和Tau聚集物的典型解剖学进展模式。
围绕错误折叠的蛋白质过程存在一系列内在机制,需要对其进行充分研究,以全面了解其在细胞中的毒性效应(Eisele,2016 ;Lewis,2016 )。这些机制包括MP的种子、聚集、繁殖和/或扩散。因此,这些MP相关机制在时间过程中的特定特征通常被称为MP动力学。识别构象疾病特异性触发事件的主要障碍在于将MP动力学在分子水平上的影响外推到更宏观的尺度上。
错误折叠蛋白的数学模型
数学模型是一种理论环境,旨在尽可能模拟和再现生物过程的真正动态。 由于微分方程式可以轻松再现多个时间交互过程的非线性和复杂动态,因此微分方程式表达的数学模型是自然之选。 在错误折叠蛋白的背景下,数学模型可以分为两大类。一类模型旨在模拟分子级过程,如种子、聚集和细胞间短程空间扩散;另一类模型则模拟更宏观的相互作用,如人脑内长程MP传播。通过这样的分类,人们可以发现微观尺度类型的模型主要是在化学动力学领域开发的。相比之下,较大尺度(即宏观)类型的模型大多是在神经影像学研究的框架内提出的。
朊病毒样动力学
由于朊病毒病也可以被视为一种MP过程(Scheckel,2018 ),因此,用于MP的开创性数学模型自然地用于模拟朊病毒复制和聚集的生物机制(Eigen,1996 )。基于常微分方程(ODE),目前被广泛接受的初始原型模型被称为核聚变模型(NPM)(Nowak,1998 ),它专注于自催化机制,旨在解释典型的朊病毒蛋白转化为传染性蛋白的过程。值得注意的是,诺瓦克等人(Nowak,1998) 意识到NPM中包含的蛋白质复制和聚集机制与流行病学模型中描述病毒动力学所采用的动力学类似。因此,现代描述MP传播的宏观模型也受到流行病学模型的启发也就不足为奇了。
通过建立朊病毒动力学NPM作为基石,多项研究(Petkova,2005 ;Frost,2009 )表明,α-突触核蛋白、tau蛋白和Aβ蛋白的纤维状聚集体在生化机制下自我繁殖,与朊病毒聚集/繁殖的描述类似。这些观察结果与多个动物体内模型相结合,为神经退行性疾病进展的“类朊病毒假说”奠定了基础(Frost,2010 )。此后,根据“类朊病毒假说”,MP的“传染性”从初始种子区域(致病蛋白浓度相对较高)传播到其他“未感染”的脑区。
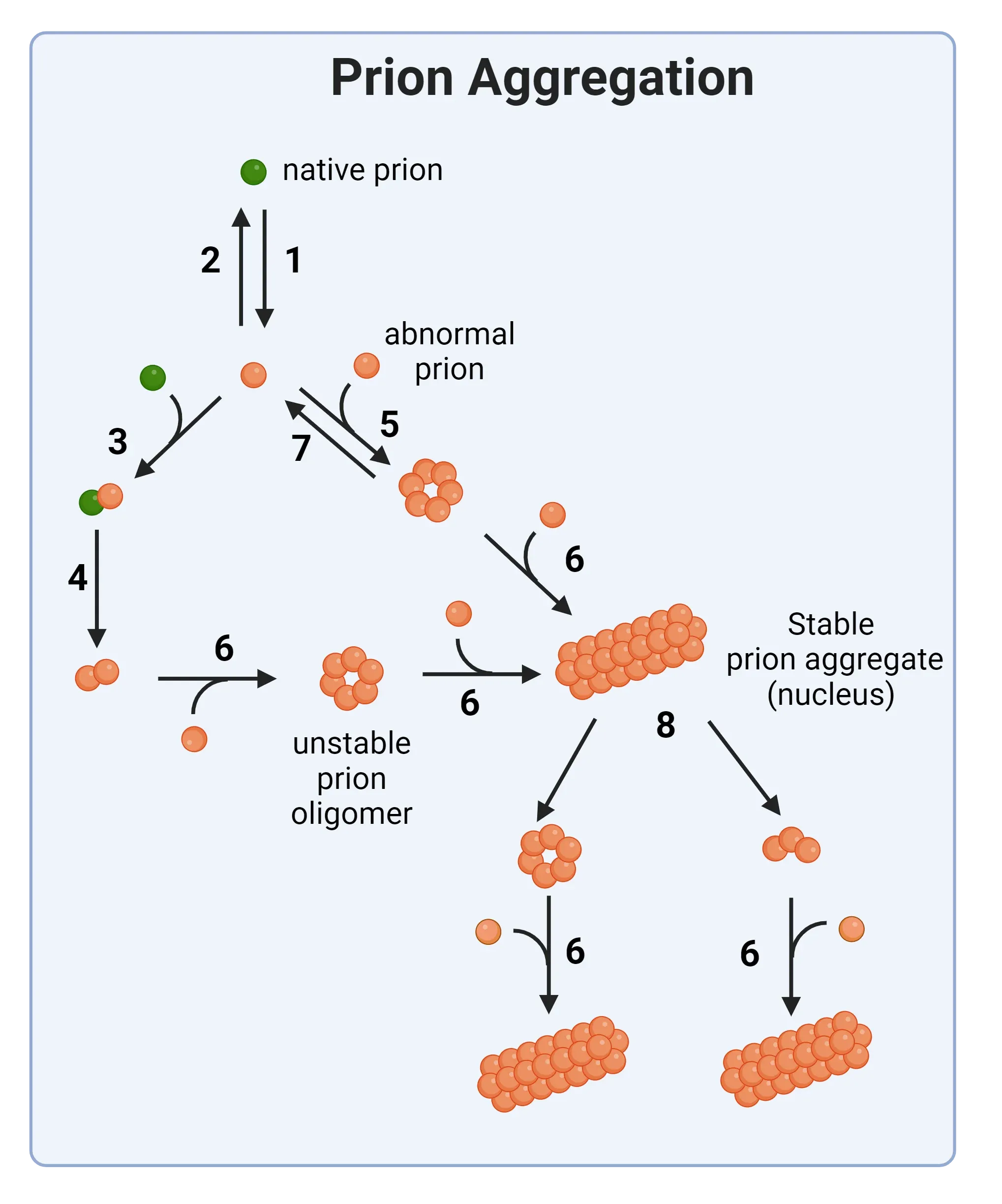
朊病毒聚集机制示意图。天然朊病毒分子发生构象变化,导致异常构象(步骤1)。此后,经过几个种子和聚集步骤,形成不稳定的朊病毒低聚体,最终产生稳定的先期聚集体。
MP传播:空间扩散
Payne等人(Payne,1998 )对MP动力学的研究取得了突破性进展,他们为先前在动物模型中观察到的外周神经系统(PNS)中传染性因子蛋白的出现与中枢神经系统(CNS)中传染性因子蛋白的出现之间的延迟现象提供了合理的解释(Scott,1992 )。因此,Payne等人(Payne,1998 )提出了一个假设,即所观察到的潜伏期更可能与邻近地区之间的传播速度有关,而不是由典型的朊病毒蛋白在特定空间位置转化为传染性蛋白的速度来解释。为了证明这一假设,需要扩展NPM的原始流行病学配置,以将空间“连接性”特征纳入朊病毒动力学的时间演化中(Stumpf,2000 )。这种扩展模型的主要基本假设是,传染性病原体蛋白通过轴突缓慢运输在空间上沿着轴突和树突传播。此外,我们自然也会假设细胞之间的传播速度取决于连接强度。
如何对错误折叠的蛋白质动力学进行数学建模?
用化学动力学模拟蛋白质聚集机理
模拟MP的最现代方法是使用化学动力学原理推导主方程,以明确解释蛋白质聚集机理中涉及的不同微观过程(Cohen,2012 ;Cohen,2013 )。其目的是用从主方程中推导出的综合速率定律来描述蛋白质聚集的动力学。
考虑到丝状生长现象以及均相成核、生长和分离过程的基本原理,Oosawa等人(Oosawa,1962 )提出了蛋白质聚集主方程模型的第一个原型。与NPM的情况一样,主方程可以通过一组无穷的常微分方程来解析描述。然而,在实践中,这样一组无穷的常微分方程可以简化为一个仅包含两个常微分方程的封闭系统,这两个常微分方程通常被称为矩方程,用于描述蛋白质聚集体数量和质量浓度的时空演化,这些量与大多数常见体外实验的测量结果直接相关。然后,通过求解矩方程,可以直接得到所需的积分速率定律(Cohen,2013 )。然而,解析求解矩方程是一项复杂的任务,特别是对于整个反应时间过程。
随后,Knowles等人(Knowles,2009 )为蛋白质形成的主方程理论做出了重大贡献,他们为适用于整个反应时间过程的集成速率定律提供了解析解。与最初的Oosawa理论一样,这些明确的集成速率定律可以描述反应时间过程对两个参数的依赖性,这两个参数可以很容易地与实验观察到的现象变量相关联(Cohen,2011 )。从实际的角度来看,拥有集成速率定律的分析公式是非常重要的,这可以允许将模型与来自不同实验条件的数据相匹配。例如,Knowles等人提出的模型(Knowles,2009 )与肽Aß40和Aß42的体外数据相匹配,从而更清楚地揭示了Aß聚合物的形成过程(Meisl,2014 ;Meisl,2016)。在过去的几年中,主方程方法和相应的综合速率定律已经过多次扩展和推广。其中最引人注目的是Cohen等人(Cohen,2014 )的研究,他们通过在主方程中加入空间传播机制,发现次级聚集路径通过扩散控制空间传播的速度。
凝聚理论
与主方程法一样,Smoluchowski方程描述的凝聚理论也涵盖了不同尺寸的粒子集合体之间的自聚合作用的一般情况 。Murphy等人 (Murphy,2000) 首次在蛋白质聚集的背景下引用了Smoluchowski方程,用于描述较短纤维通过端到端聚集产生的轴向伸长。同样,Craft等人(Craft,2005 )使用Smoluchowski的方程提出了聚合模型,其中成核过程隐含地包含在小尺寸聚合物(例如单体、胶束、丝状物)的聚集机理中。
Achdou等人(Achdou,2013 )在蛋白质聚集和传播机制建模领域取得了重大突破。Achdou等人(Achdou,2013) 没有使用矩量方程,而是将无穷大的常微分方程组截断为足够大的N。这样,截断数N对应的方程应该能够描述由超过N个单体组成的全部蛋白质组装体的总时间演化。此外,Achdou等人(Achdou,2013 )发现,斯莫卢霍夫斯基方程还可以提供一个直接框架,用于整合空间传播过程。
现代成像技术的发展要求能够对比微观尺度更大的尺度进行建模。为此,Bertsch等人提出了一个大型模型(Bertsch,2017),将一组截断的斯莫卢霍夫斯基方程与动力学型传输方程耦合,后者模拟了神经元对神经元朊病毒样传播的神经元损伤扩散。这种建模能够整合在同一空间域内演化的两种不同时间尺度:一种为微观过程的快速时间尺度(例如分钟、小时)(通过Smoluchowski’s方程),另一种为解释人类AD观察进展的慢速时间尺度(例如月、年)(通过传递方程)。
用网络方法模拟跨区域传播
在大脑等大型空间域中,用均质扩散过程模拟错误折叠蛋白的空间扩散并不现实。与均质扩散传播相反,网络方法模拟了高度连接的神经元集群内快速感染传播,以及通过长距离连接传播到其他集群的过程(Matthäus,2006 )。因此,通过使用离散易感-感染(SI)流行病学模型,Matthäus等人(Matthäus,2006 )描述了朊病毒蛋白感染沿着相互连接的神经元网络传播的特征。更重要的是,这种基于网络的数学框架可以很容易地适应描述更大规模的场景,其中网络节点代表覆盖大空间域的遥远区域。
在这方面,Raj等人(Raj,2012 )提出了宏观网络扩散模型(NDM),其中从特定脑区到任何其他区域的MP传入神经的数量仅取决于两个区域中的疾病浓度因子以及它们之间的解剖连接强度。考虑到更现实的场景,Iturria-Medina等人(Iturria-Medina,2014 )提出了流行病传播模型(ESM),该模型同时考虑了区域产生/清除MP的能力以及大脑解剖网络的拓扑信息。当使用淀粉样蛋白PET数据对晚发性AD进行研究时,该模型能够再现个体水平的淀粉样蛋白沉积模式。由于存在详细的病理机制和AD进展的假设,人们开始考虑采用一种更综合的多因素建模方法,这种方法超越了错误折叠蛋白的形成和传播(Edelstein-Keshet,2002 ;Luca,2003 )。
治疗干预的理论模型如何应用?
通常,蛋白质聚集的数学模型被用作体外治疗干预的替代品。一般思路是模拟治疗干预(例如药物、抗体、分子伴侣)如何抑制某些微观聚集过程(Arosio,2014 ;Nasica-Labouze,2015 ;Kulenkampff,2021 ;Ghosh,2023 )。然后,在自然条件和抑制条件下,可以估算和比较蛋白质聚集的动力学速率。首先假设适当的动力学速率发生假设变化(例如通过处理),然后将这些修改后的速率代入原始模型,以模拟处理后的状态。
监测动力学速率与假设药物剂量之间的关系,有助于将体外环境中较小的药物剂量推断为更接近体内条件的剂量(Masel,2000 )。在开创性的研究中,Masel等人(Masel,2000 )使用数学模型模拟淀粉样蛋白传播的抑制,主要采用三种方法:(1)降低未折叠蛋白的有效单体浓度;(2)阻断不断增长的高分子末端;(3)提高高分子的断裂率。他们发现,采用第二种策略的治疗效果会很好,而其他策略可能无效,甚至在低剂量下会加速淀粉样蛋白的形成过程。
通过三种不同的策略模拟抑制淀粉样蛋白的聚集/扩散:(1)降低有效单体浓度;(2)阻断不断增长的聚合物末端;(3)增加聚合物的断裂率。
另一个例子是,在淀粉样β蛋白的背景下,Craft等人(Craft,2005 )在聚合率和总Aß负荷之间建立了非线性关系。由于聚合率取决于四种不同的动力学速率,因此可以在此背景下测试几种治疗策略:(1)降低Aß单体生产率;(2)增强断裂;(3)提高清除率(即降解率);(4)降低伸长率。通过对上述情况的模拟,Craft等人(Craft,2005 )得出结论:与基于聚合物断裂增强剂的药物相比,基于清除率增强剂的药物可能更有效地降低总Aß负担。
最近,Arosio等人(Arosio,2016 )使用化学动力学方法模拟分子伴侣与不同蛋白质之间的相互作用。主要思路是确定分子伴侣与特定蛋白质结合后,哪些微观反应步骤(即初级成核、伸长、断裂和次级成核)受到干扰。Arosio等人(Arosio,2016 )使用主方程方法,比较了不同浓度分子伴侣存在和不存在情况下的估计动力学速率常数。对于Aß42蛋白,此类分析表明,名为DNAJB6的分子伴侣的作用会抑制初级成核过程。相比之下,另一种分子伴侣(名为proSP-C Brichos)的存在会降低二级成核速率。
作为进一步的例子,Habchi等人(Habchi,2016 )使用化学动力学方法开发了一种合理的药物发现策略,并报告了一种抗癌药物(称为bexarotene)干扰了Aß42聚集中的初级成核步骤,延迟了有毒低聚物的形成,并完全抑制了Aß42的沉积。这一总体框架形成了一种系统的药物发现策略,旨在识别各种小分子(Habchi,2017 )和抗体(Aprile,2017 ),这些小分子和抗体不仅针对聚集的起始阶段,还针对导致毒性Aß42低聚物增殖的二次成核步骤。
化学动力学和蛋白质聚集领域的最新进展无疑重新激发了人们对治疗干预建模的兴趣,将其视为发现MP相关疾病新潜在治疗药物的有用工具(Linse,2020 ;Kulenkampff,2021 ;Ghosh,2023 )。
我们的团队很乐意回答有关神经疾病中蛋白质折叠异常建模的任何问题,或提供有关我们其他成像服务的具体信息。
相关内容
神经影像学在神经疾病临床试验中的最佳实践的最新信息。
亨廷顿病临床试验中的正电子发射断层成像
PET成像生物标记在亨廷顿病(HD)临床试验中的应用概述。
MCI和阿尔茨海默氏症患者脑部Tau蛋白PET的纵向变化
轻度认知障碍(MCI)和阿尔茨海默氏症(AD)患者对Tau PET示踪剂吸收和结合的自然变化历史概述。
通过生物标记区分CBD与其他Tau蛋白病
大脑成像生物标志物概述,用于识别皮质基底变性(CBD)患者,以及其在疾病治疗临床试验中的应用。
额颞叶痴呆症和临床试验中的神经影像学
核磁共振成像和正电子发射断层扫描成像生物标记物在了解额颞叶痴呆(FTD)变体方面的效用,以及它们在FTD临床试验中的终点用途。