Spinocerebellar Ataxia Clinical Trials & Imaging Biomarkers
An overview of structural MRI, DTI and MRS imaging biomarkers to monitor disease progression in spinocerebellar ataxia clinical trials.
What are the clinical features and genetic causes of SCA?
Spinocerebellar ataxia (SCA) refers to a heterogenous group of over 40 rare, autosomal dominant neurodegenerative disorders. These disorders are characterized by progressive degeneration of the cerebellum, brainstem, and spinal cord. Ataxia, which is the most common and often the earliest clinical symptom is typically assessed using the Scale for the Assessment and Rating of Ataxia (SARA), which measures the severity of the condition (Schmitz-Hübsch, 2006). Ataxia is typically accompanied by gait instability, limb incoordination, oculomotor abnormalities, and dysarthria (difficulty speaking) (Rossi, 2014; Cui, 2024). Patients may also experience a pre-ataxic stage, which can last for several years before noticeable ataxic symptoms appear. During this phase, subtle coordination deficits and brain abnormalities gradually develop (Brooker, 2021; Cui, 2024). As the disease progresses, patients may also experience non-ataxic symptoms, such as cognitive impairments, spasticity, rigidity, and dystonia (Brooker, 2021). These symptoms can result in significant morbidity, with many patients eventually becoming wheelchair-bound and experience premature death.
The classification of SCAs follows a numerical system based on the order in which the genetic mutations were discovered. The most common forms are those associated with polyglutamine (polyQ)-encoding CAG repeat expansions, which include SCA1, SCA2, SCA3, SCA6, SCA7, and SCA17, which are the most extensively studied as they account for over half of all known cases (Brooker, 2021). SCA3 (also known as Machado-Joseph disease), SCA2, and SCA6 are the most prevalent subtypes (Afonso-Reis, 2021). In contrast, other SCAs result from noncoding repeat expansions, including SCA8, SCA10, SCA12, SCA27B, SCA31, SCA36, and SCA37. The remainder of the SCAs are linked to point mutations in specific genes (Cui, 2024). SCA typically manifests during early adulthood, often around the fourth decade of life, though the age of onset can vary widely (Rossi, 2014; Afonso-Reis, 2021). For SCAs caused by repeat expansions, a critical factor influencing the age of onset and severity of symptoms is the length of the repeat expansion, with longer repeats generally associated with earlier onset and more severe progression (Brooker, 2021).
Developing effective treatments for SCA is particularly challenging due to the diversity in their underlying pathogenesis. Moreover, clinical phenotypes can overlap among different SCA subtypes, making genetic testing essential for a definitive diagnosis. Furthermore, there are varying rates of progression across different genotypes. For instance, ataxia tends to progress more rapidly in SCA1, at an intermediate rate in SCA2 and SCA3, and more slowly in SCA6 (Diallo, 2021). Additionally, the rarity of SCAs, affecting approximately 1-5 individuals per 100,000 people, also complicates patient recruitment for clinical trials. However, research on other polyglutamine repeat disorders, particularly Huntington’s disease (HD), has provided valuable insights into treatment approaches for polyQ SCAs. Current research is focused on exploring novel therapeutic strategies, particularly during the pre-ataxic stage, when treatments could potentially delay or even prevent the onset of more severe symptoms. Thus, identifying sensitive neuroimaging biomarkers that can detect brain changes before clinical symptoms manifest is essential for early diagnosis and for evaluating the efficacy of disease-modifying treatments in clinical trials (Cui, 2024).
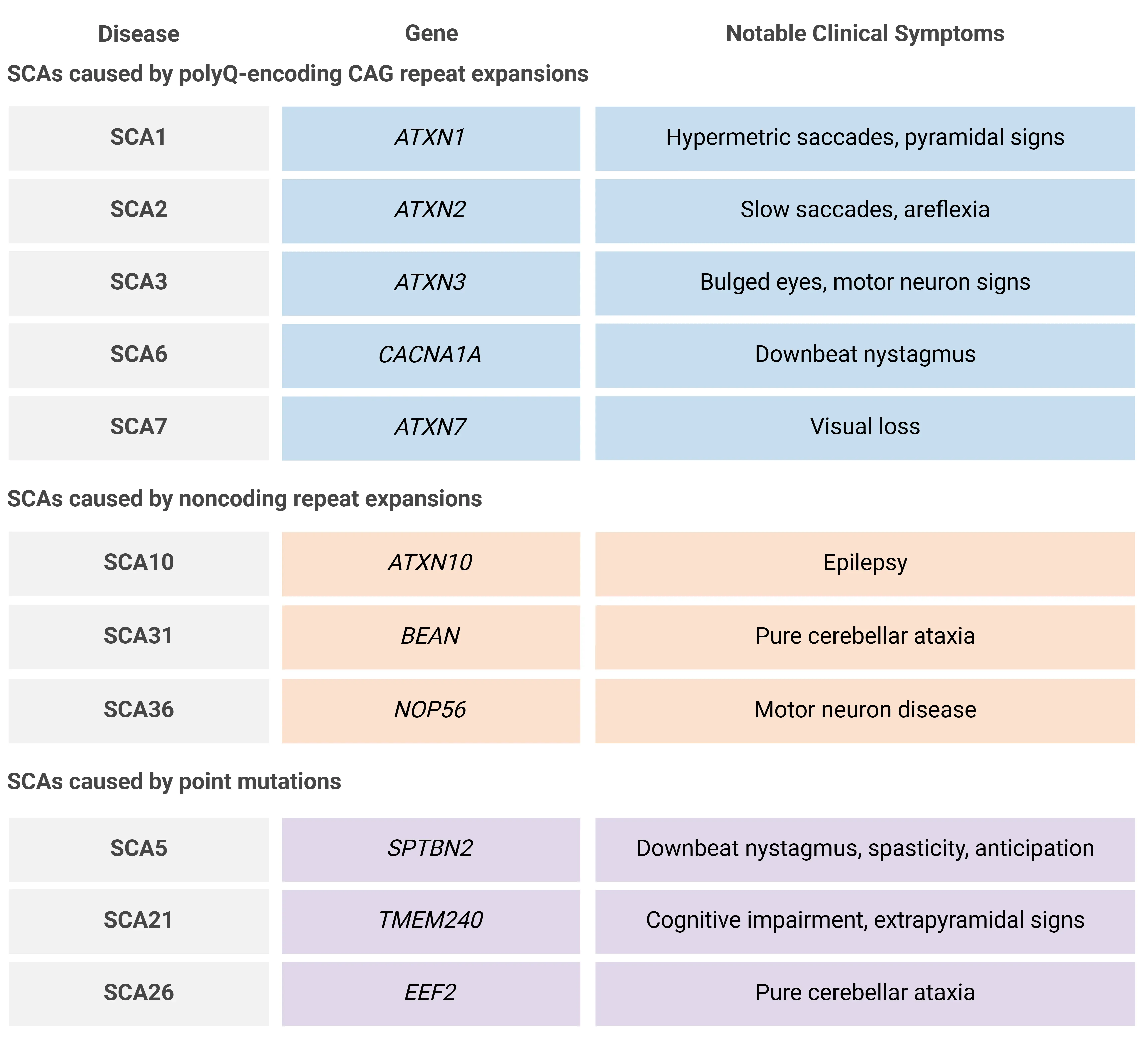
Table representing a small fraction of the over 40 known SCAs, including a subset of those caused by polyQ-encoding CAG repeat expansions, noncoding repeat expansions, and point mutations, along with their associated genes and notable clinical symptoms. While ataxia is the most common symptom, clinical manifestations vary across subtypes. Adapted from Cui et al. (Cui, 2024) under the Creative Commons Attribution License.
Which neuroimaging biomarkers are most effective for tracking SCA progression?
Structural Magnetic Resonance Imaging (MRI)
Structural MRI is a key tool for assessing brain region involvement in SCA, revealing that the extent of cerebellar, spinal cord, and brainstem atrophy varies across different SCA subtypes (Iwabuchi, 2022). A study comparing early-stage SCA1, SCA2, SCA3, and SCA7 revealed significant reductions in cerebellar and pons volumes in all groups over 24 months (Adanyeguh, 2018). Longitudinal analysis showed that SCA patients experienced faster atrophy rates in these regions compared to controls, with the exception of SCA7, where cerebellar atrophy rate did not significantly differ from controls. Volume differences were also observed in the pallidum, medulla, and midbrain at both baseline and follow-up (Adanyeguh, 2018).
Volume changes in pre-ataxic SCA patients, offer valuable insights into the progression from pre-ataxic to ataxic stages. In SCA2, brainstem volumes, particularly in the pontine region, are reduced in both ataxic and pre-ataxic patients (Reetz, 2018). In SCA17, atrophy has been observed in both the cerebellum and caudate nucleus in both pre-ataxic and ataxic patients (Brockmann, 2012). These findings hold potential for distinguishing SCA stages, and serve as promising biomarkers for monitoring disease progression.
In early ataxic and pre-ataxic stages of SCA3, studies report differing results. Faber et al. (Faber, 2021) found that cerebellar volume loss was less pronounced than in the spinal cord and brainstem, with a steady decline in pons volume, which was most significant at the onset of ataxia. In contrast, Rezende et al. (Rezende, 2024) observed significant changes in right cerebellar volume, with high effect sizes even at early stages, but no changes in pons volume. These contrasting neuroimaging findings may reflect variability in SCA3 disease progression and symptom severity, underscoring the importance of proper patient stratification in clinical trials.
Diffusion Tensor Imaging (DTI)
White matter abnormalities detected by DTI accompany the structural changes observed in SCA. In early-stage SCA1, SCA2, and SCA3, decreased fractional anisotropy (FA) and elevated radial diffusivity (RD) are seen across multiple tracts when compared to controls (Adanyeguh, 2018). These changes are most pronounced in the cerebellar and cerebral peduncles, and pontine crossing tracts. Abnormalities in the corticospinal tract, corona radiata, and internal capsule are also found in SCA1 and SCA2, with SCA2 showing reduced FA in the corpus callosum (Adanyeguh, 2018). In contrast, elevated RD, but no changes in FA, are observed in SCA7, while no changes in the corpus callosum, internal capsule, or corona radiata are found in SCA3 (Adanyeguh, 2018). However, other studies have reported reduced FA and increased RD in early-stage SCA3, specifically in the right corona radiata and right superior longitudinal fasciculus, highlighting opposing findings for SCA3 in the corona radiata (Rezende, 2024). These opposing findings for SCA3 in the corona radiata may be attributed to differences in patient populations or disease stages, indicating the need for further investigation into these conflicting results.
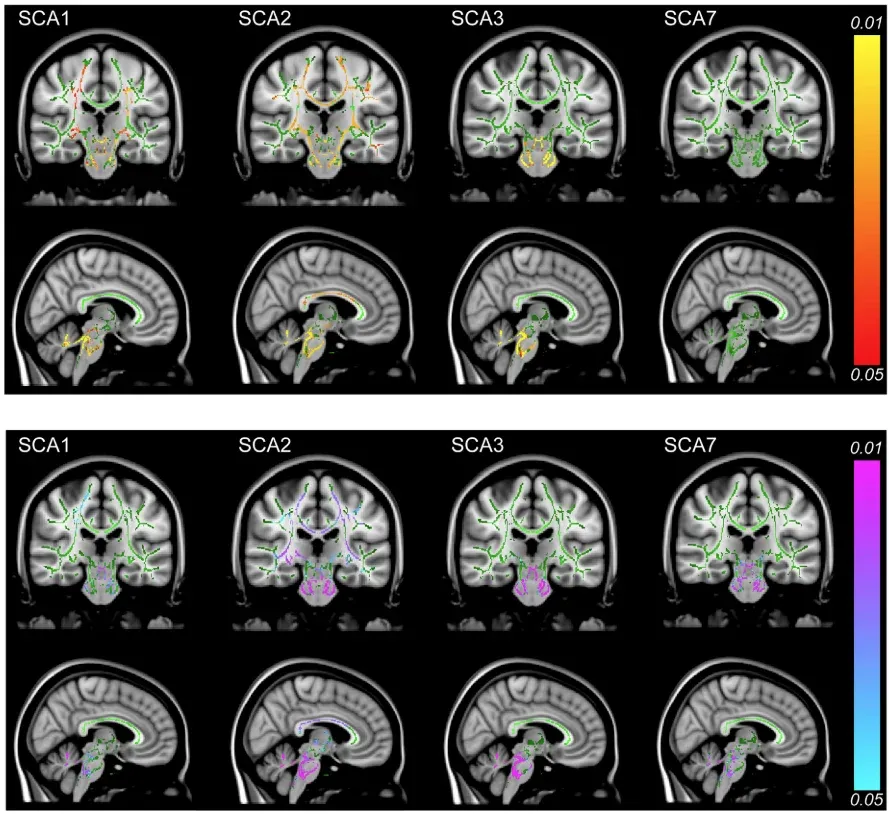
DTI shows lower FA in SCA1, SCA2, and SCA3 (top panel) and higher RD across all groups (bottom panel). Changes are most pronounced in the cerebellum, cerebral peduncles, and pontine crossing tracts, with additional involvement of the corticospinal tract, corona radiata, internal capsule (SCA1, SCA2), and corpus callosum (SCA2). Red-yellow areas indicate decreased FA, and blue-pink regions show increased RD. Figure reproduced from Adanyeguh et al. (Adanyeguh, 2018) under the Creative Commons Attribution License.
Magnetic Resonance Spectroscopy (MRS)
MRS is a sensitive tool for detecting neurochemical changes, even in pre-ataxic SCA, making it useful for tracking disease progression (Brooker, 2021; Chen, 2021). In early-to-moderate stage SCA1 and SCA2, total N-acetylaspartate (tNAA) and glutamate are reduced in the cerebellum and pons compared to controls, while myo-inositol and total creatine (tCr) are elevated (Öz, 2010; 2011). In SCA1, the tNAA/myo-inositol ratio in the cerebellum and pons differs from controls with high sensitivity and specificity (Öz, 2010). Reductions in NAA/Cr and NAA/choline ratios in the cerebellum are observed in SCA1, SCA2, SCA3, SCA6, and SCA17 (Chen, 2021). However, SCA6 shows fewer neurochemical alterations compared to controls, with less involvement of the pons and a higher cerebellar NAA/Cr ratio than SCA1 and SCA2 (Öz, 2011). These findings highlight MRS as a promising tool for detecting neurochemical changes in SCA. Additionally, MRS plays a crucial role in differentiating between various SCA subtypes and distinguishing them from controls, thereby aiding in patient stratification and serving as a valuable biomarker in clinical trials.
Clinical Correlations
Neuroimaging findings correlate with clinical measures, offering insights into disease progression and patient outcomes. Reduced cerebellar volume is closely correlated with ataxia severity, as measured through region of interest (ROI)-based analysis and voxel-based morphometry (VBM) in SCA1, SCA2, SCA6, and SCA17 (Chen, 2021). The SARA score correlates with volumes of the cerebellum, brainstem, caudate, spinal cord, cerebellar white matter, as well as with DTI metrics across multiple tracts (Adanyeguh, 2018; Chen, 2021). Importantly, clinical scores exhibit smaller longitudinal effect sizes compared to the large effect sizes seen with volumetric changes, suggesting that MRI biomarkers may provide more sensitive measures of disease progression, making them valuable tools in clinical trials (Adanyeguh, 2018; Faber, 2021).
Additionally, MRS findings have been shown to correlate with clinical outcomes. In SCA1, levels of tNAA, myo-inositol, and glutamate in the cerebellum, and tNAA and myo-inositol in the pons, correlate with the SARA score (Öz, 2010). The NAA/Cr ratio in the cerebellar vermis correlates with the SARA score in SCA2 and SCA3, while the NAA/Cr ratio in the right cerebellar hemisphere shows a stronger correlation with the SARA score in SCA6 (Wang, 2012). These findings highlight that different MRS measures correlate with clinical scores in distinct ways across SCA subtypes, offering insights into how neurochemical changes may reflect disease progression in each subtype. Given the complex nature of SCA, a multimodal biomarker approach is recommended to improve the accuracy and utility of disease progression monitoring (Adanyeguh, 2018; Chen, 2021).
What treatment approaches are current clinical trials investigating for SCA?
Currently, no disease-modifying treatments exist for SCA, although several approaches aimed at slowing disease progression are under investigation. The main challenges to conducting clinical trials arise from the rarity of SCA, their variable severity and progression, and clinical and genetic heterogeneity. To address these complexities, researchers are exploring a variety of therapeutic strategies, including stem cell technologies, RNA interference (RNAi), antisense oligonucleotides (ASOs), CRISPR-based approaches, autophagy modulators, and pharmacological interventions, among others (Kwei, 2020; Brooker, 2021; Ghanekar, 2022; Correia, 2023).
Ongoing clinical trials are investigating Stemchymal®, a stem cell technology designed to slow disease progression (NCT06397274, NCT03378414), glutamate modulators such as troriluzole (NCT06529146, NCT03701399), and RNAi therapies like ARO-ATXN2 (NCT06672445). ASOs, including VO659, are also being investigated for both SCA and HD (NCT05822908). Additionally, several natural history studies focused on various SCA subtypes are currently underway, which have the potential to provide valuable insights into disease progression, and inform the development of targeted therapies (NCT02741440, NCT02440763, NCT01060371, NCT06472557). Neuroimaging biomarkers are also being evaluated in clinical trials and could play a crucial role in offering objective measures of disease severity, monitoring treatment response, and enhancing the design of future therapeutic interventions (NCT05160883, NCT04297891).
Given the shared pathogenesis among SCAs, it has been proposed that disease-modifying therapies could be developed for multiple subtypes (Cui, 2024). However, the distinct underlying proteins for each SCA subtype present a challenge to this approach, suggesting that personalized treatments tailored to the genetic causes of individual SCAs may be necessary (Cui, 2024).
When designing clinical trials, several factors must be considered, such as the age of onset, repeat length, and age at trial inclusion, as these factors are important for patient stratification and predicting disease severity. For polyQ SCAs, CAG repeat length is a critical determinant of disease severity (Brooker, 2021). Effective recruitment for trials also requires collaboration and the establishment of patient registries. In the U.S., the Clinical Research Consortium for Spinocerebellar Ataxias (CRC-SCA) plays a central role in advancing research, while global initiatives like SCA Global aim to accelerate biomarker discovery and treatment development by standardizing research methods and expanding efforts internationally (Brooker, 2021).
Our team would be happy to answer any questions about Spinocerebellar Ataxia Clinical Trials & Imaging Biomarkers or provide specific information about our other Imaging services.
Discover more about our Imaging Services
Related Content
Up-to-date information on best practices related to the use of neuroimaging in clinical trials of neurological diseases.
Imaging Biomarkers for Friedreich’s Ataxia Clinical Trials
An overview of the use of MRI and DTI imaging biomarkers for Friedreich’s ataxia (FRDA) in research studies & multi-center clinical trials.
MRI Biomarkers in Clinical Trials of Huntington’s Disease
An overview of the use of structural and diffusion MRI imaging biomarkers for Huntington’s disease (HD) clinical trials.
PET Imaging in Huntington’s Disease Clinical Trials
An overview of the use of PET imaging biomarkers for Huntington’s disease (HD) clinical trials.
MRI in Clinical Trials of Multiple System Atrophy (MSA)
This resource provides an overview of the utility of volumetric MRI and diffusion-weighted imaging (DWI) as biomarkers in research studies of MSA.