MRI Biomarkers in Clinical Trials of Huntington’s Disease
An overview of the use of structural and diffusion MRI imaging biomarkers for Huntington’s disease (HD) clinical trials.
What is the clinical presentation and genetic basis of HD?
Clinical Presentation
Huntington’s disease (HD) is a rare, progressive neurodegenerative disorder inherited in an autosomal dominant pattern, typically presenting with adult-onset symptoms between the ages of 35 and 40. Its incidence is approximately 4-10 per 100,000 in individuals of European descent (Klöppel, 2009). It is primarily characterized by the degeneration of medium spiny neurons (MSNs) in the striatum. Neuroimaging studies reveal early atrophy in the caudate and putamen, along with a gradual loss of cerebral white matter, which can be detected years before clinical symptoms manifest. As the disease advances, individuals experience a progressive decline in motor control, cognitive function, and neuropsychiatric health.
Motor symptoms in HD involve both involuntary and voluntary movement difficulties. Involuntary movements include chorea (irregular, jerky movements), dystonia (muscle contractions), and rigidity. Voluntary motor control also deteriorates, leading to symptoms such as dysarthria (difficulty with speech), dysphagia (difficulty swallowing), and akinesia (a reduced ability to initiate movement) (Jiang, 2023). Cognitive decline is marked by deficits in executive functions, particularly difficulties in goal-directed behavior, along with impairments in verbal learning, visuospatial abilities, and ultimately, severe memory loss. Neuropsychiatric symptoms are also common and may include anxiety, irritability, depression, obsessive-compulsive behaviours, aggression, apathy, and psychosis (Jiang, 2023). Additionally, individuals with HD often experience unintended weight loss, sleep disturbances, and circadian rhythm disruptions.
The typical onset of HD occurs between the ages of 35 and 40, although around 5% of cases present as juvenile Huntington’s disease (JHD), with onset before the age of 20. The disease is commonly described in three stages. The first stage, known as the presymptomatic (or premanifest) stage, is marked by the absence of any noticeable clinical abnormalities. The next phase is the prodromal phase, in which subtle changes in motor function, cognition, and behavior begin to appear. The final stage is the manifest stage, during which symptoms become pronounced, and a formal diagnosis of HD is made (Jiang, 2023).
Diagnosing HD involves a combination of genetic testing, family history assessment, and clinical evaluation. The Unified Huntington’s Disease Rating Scale (UHDRS) is frequently used to assess motor symptoms, while neuroimaging methods, such as magnetic resonance imaging (MRI), help exclude other conditions and monitor disease progression. One key neuroimaging finding is caudate atrophy, which is particularly sensitive for tracking the progression of the disease and has become an important biomarker in clinical trials aimed at evaluating new treatments (Hobbs, 2024).
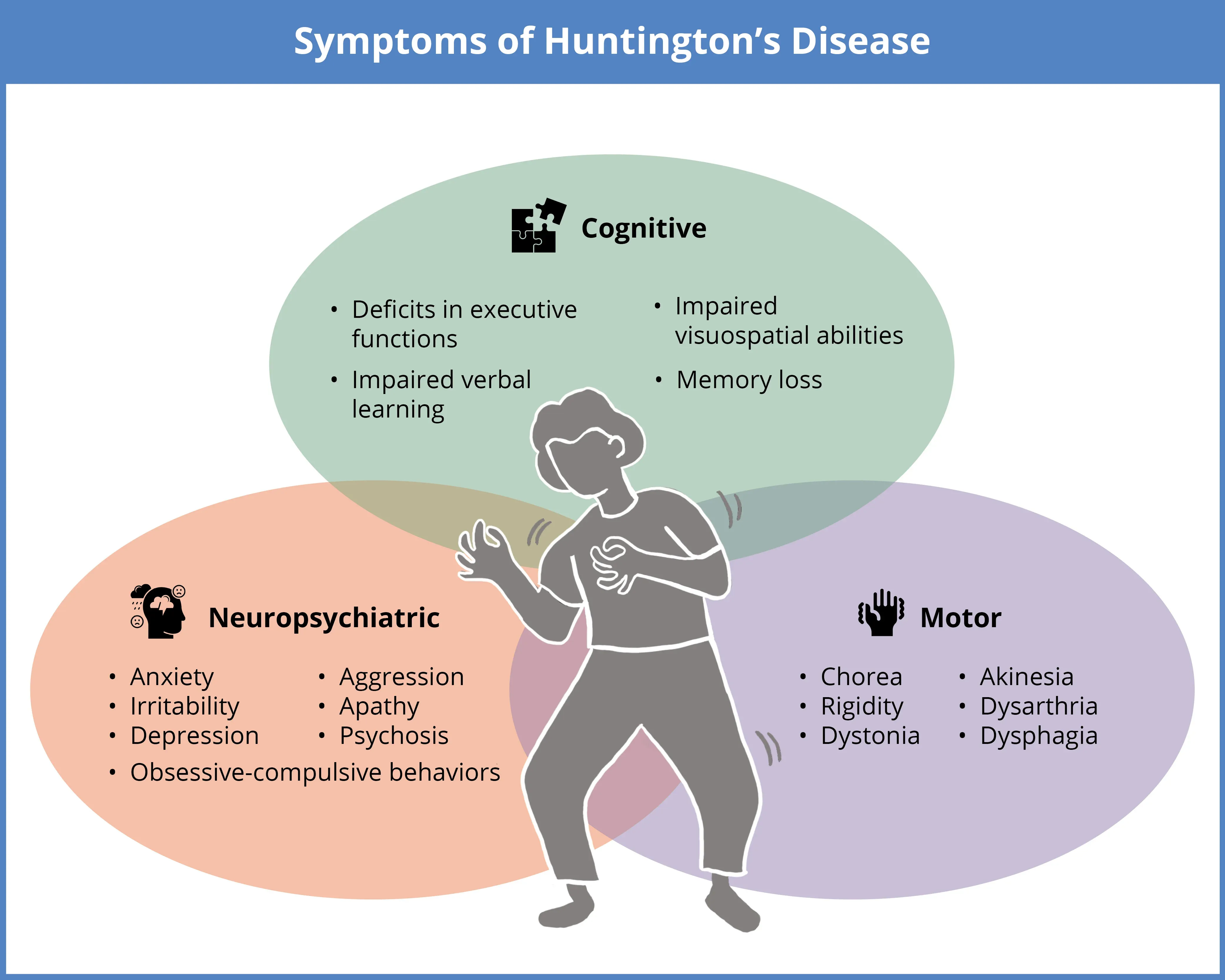
Huntington’s disease (HD) is a rare, progressive neurodegenerative disorder characterized by a triad of symptoms, which may include cognitive decline (executive function deficits, impairments in verbal learning and visuospatial skills, and memory loss), neuropsychiatric changes (anxiety, irritability, depression, obsessive-compulsive behaviors, aggression, apathy, and psychosis), and motor dysfunction (chorea, rigidity, dystonia, akinesia, dysarthria, and dysphagia).
 Click to copy link
Click to copy link
Genetic Basis
HD results from a mutation in the HTT gene, which encodes the huntingtin protein. The mutation consists of an abnormal expansion of the CAG trinucleotide repeat within the first exon of the HTT gene on chromosome 4. While the normal gene contains 10 to 35 repeats, in HD, the number of repeats surpasses a critical threshold, resulting in the production of a toxic form of huntingtin protein, referred to as mutant huntingtin (mHTT) (Reiner, 2011). The accumulation of mHTT disrupts cellular functions, leading to neuronal damage. Larger expansions of the CAG repeat correlate with an earlier onset of the disease and faster progression, with those exceeding 39 repeats considered fully penetrant (Reiner, 2011; Wijeratne, 2021). JHD, often associated with CAG repeat expansions greater than 60, typically presents with symptoms such as rigidity, seizures, and behavioral changes, in contrast to adult-onset HD, where chorea is more common (Reiner, 2011). HD is part of a broader class of disorders involving polyglutamine repeat expansions, including spinocerebellar ataxias.
The CAG and age product (CAP) score is an important tool used to assess the severity of mHTT exposure and can predict the pathology observed during autopsy. This scoring system helps estimate disease progression and has been shown to correlate with clinical outcomes (Zhang, 2011).
With the discovery of the HTT gene mutation, HD is one of the few neurodegenerative disorders for which predictive genetic testing is available. This testing enables individuals with a family history of HD to determine whether they carry the genetic mutation or have presymptomatic HD (Andica, 2020). The ability to identify HD-gene mutation carriers early provides significant benefits, including the opportunity to participate in clinical trials focused on disease-modifying therapies, including potential preventative treatments.
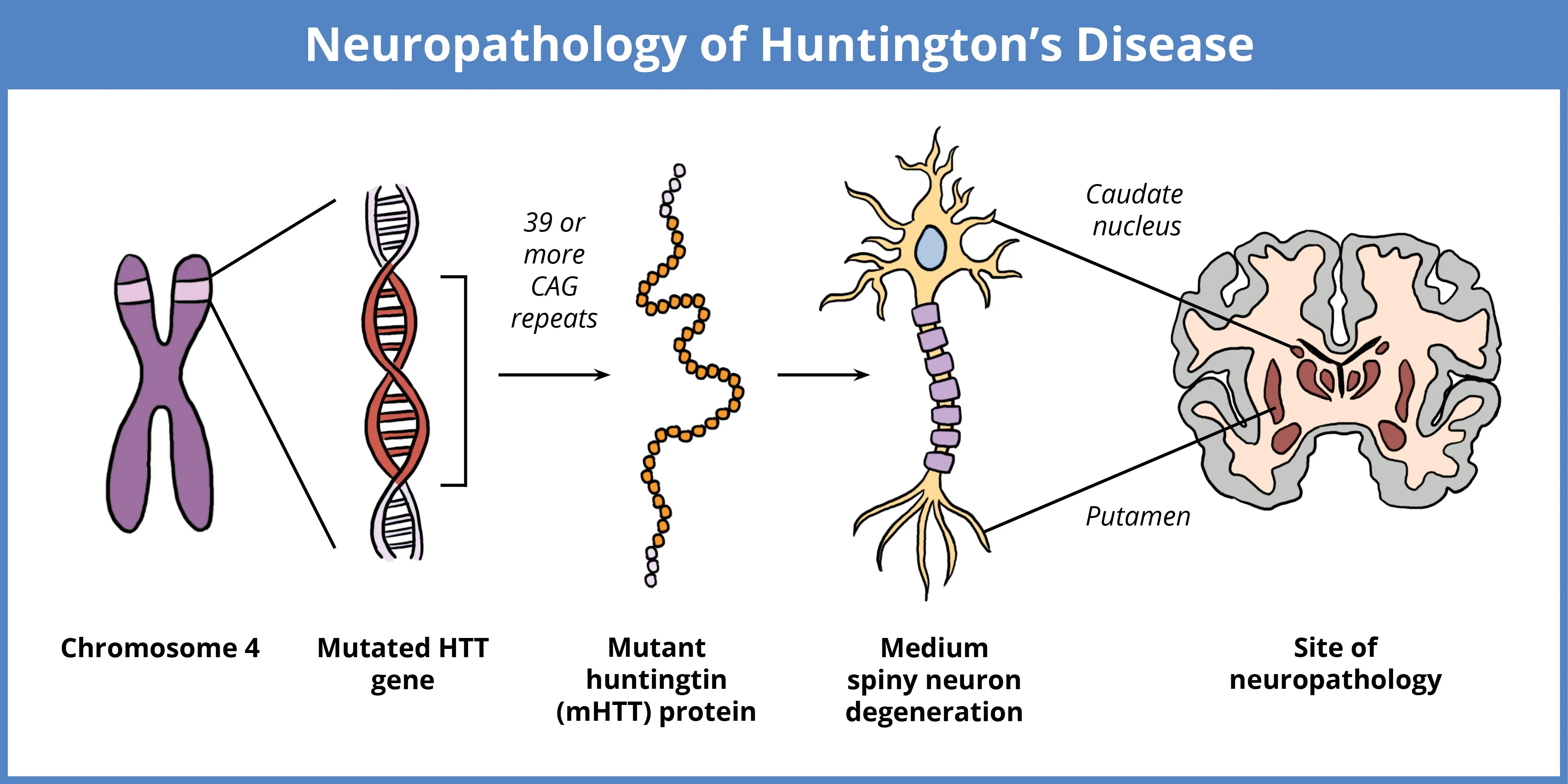
HD is caused by a mutation in the HTT gene located on chromosome 4, characterized by an expanded CAG repeat. While normal individuals have 10 to 35 repeats, expansions beyond 39 repeats results in the production of the toxic mutant huntingtin protein (mHTT). Larger repeats are linked to earlier disease onset. The accumulation of mHTT, particularly in medium spiny neurons (MSNs) of the striatum, causes neurodegeneration, resulting in motor, cognitive, and neuropsychiatric symptoms.
The clinical manifestation of HD varies significantly, with differences in age of onset, symptom presentation, disease progression, and CAG repeat length contributing to the complexity of the disorder (Cao, 2024). This variability underscores the need for reliable biomarkers to enable early diagnosis and provide accurate, effective means of evaluating treatment outcomes. In clinical trials, integrating data from multiple biomarkers is crucial to reliably stratify participants into subgroups and enhance the precision of therapeutic approaches (Wijeratne, 2018).
Which MRI biomarkers are effective for tracking disease progression in HD?
Neuropathological changes in HD are most prominent in the striatum, including the caudate and putamen. The primary pathological feature is the degeneration of GABAergic MSNs, which are critical for motor control and cognitive function. As HD progresses, neurodegeneration also affects the cerebral cortex. Both structural and diffusion MRI studies have shown that brain abnormalities can be detected well before the onset of clinical symptoms. Structural MRI, for example, reveals early changes like striatal atrophy, while diffusion MRI identifies microstructural alterations in specific white matter tracts (Reiner, 2011; Estevez-Fraga, 2023). These imaging techniques play a critical role in the early detection of HD and provide important insights into the disease’s progression.
Structural MRI Biomarkers
Volumetric MRI is an important tool for monitoring the progression of HD. Striatal atrophy is considered one of the most reliable and sensitive biomarkers, as it strongly correlates with CAG repeat length and the age at which clinical symptoms manifest (Fazio, 2018; Kinnunen, 2021). Studies have demonstrated that striatal atrophy can be detected up to 23 years before the onset of motor symptoms, making it a valuable biomarker for both disease monitoring and clinical trials aimed at evaluating therapeutic interventions (Klöppel, 2009; Hobbs, 2024). Early atrophy in the caudate and putamen is associated with some of largest effect sizes among imaging biomarkers (Hobbs, 2015). A study examining the progression of the disease using brain volume biomarkers identifies the putamen and caudate as the first areas to exhibit atrophy (Wijeratne, 2018). As HD progresses, atrophy spreads to other brain regions. Cortical thinning, particularly in areas such as the occipital, motor, dorsomedial prefrontal, and parietal cortices, often occurs prior to the onset of motor symptoms (Kinnunen, 2021). Additionally, cerebral white matter volume begins to decline early in the disease and continues to deteriorate as the disease progresses (Kinnunen, 2021). In contrast, atrophy in regions like the thalamus and hippocampus is less pronounced (Fazio, 2018).
Diffusion MRI Biomarkers
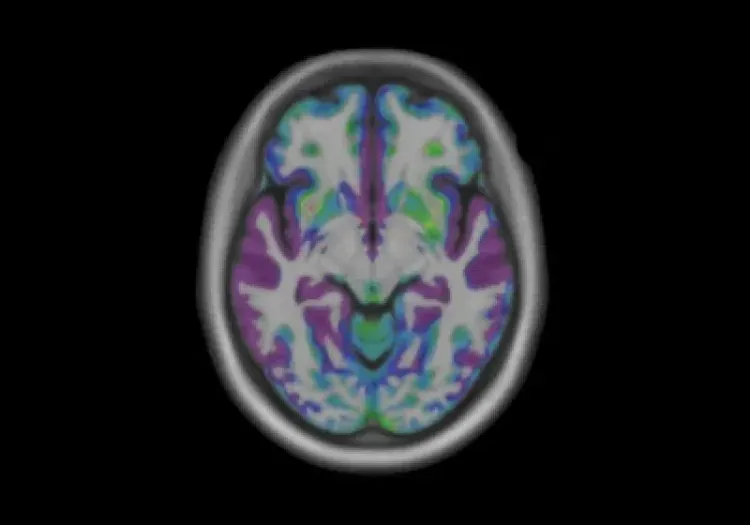
Diffusion-weighted imaging (DWI) is particularly useful for detecting early microstructural changes in HD. Longitudinal studies show reductions in fractional anisotropy (FA) in specific white matter tracts, such as the left superior longitudinal fasciculus in premanifest HD (Estevez-Fraga, 2023). Moreover, decreased FA in the corpus callosum can be observed several years before the onset of motor symptoms, highlighting the potential of diffusion MRI as an early biomarker for HD (Hobbs, 2012; Fazio, 2018). These FA reductions are accompanied by increases in mean diffusivity (MD) in the superior longitudinal fascicles, splenium of the corpus callosum, corona radiata, and external capsules, suggesting a specific pattern of white matter degeneration in HD (Estevez-Fraga, 2023). In premanifest HD-gene mutation carriers, increased MD is also observed within the basal ganglia and thalamus (Fazio, 2018). As the disease progresses to its manifest stage, patients exhibit more widespread degeneration, with extensive reductions in FA and increases in MD observed in both deep and superficial brain structures (Estevez-Fraga, 2020). Longitudinal diffusion studies show increased MD, axial diffusivity (AD), and radial diffusivity (RD) in the corpus callosum, corona radiata, fornix, frontal subcortical white matter, and external capsules (Estevez-Fraga, 2023). Interestingly, some studies have reported inconsistent findings in diffusion metrics within gray matter structures, as well as unexpected increases in both FA and MD in striatal regions–results that warrant further investigation (Klöppel, 2009; Hobbs, 2012; Fazio, 2018; Hobbs, 2024).
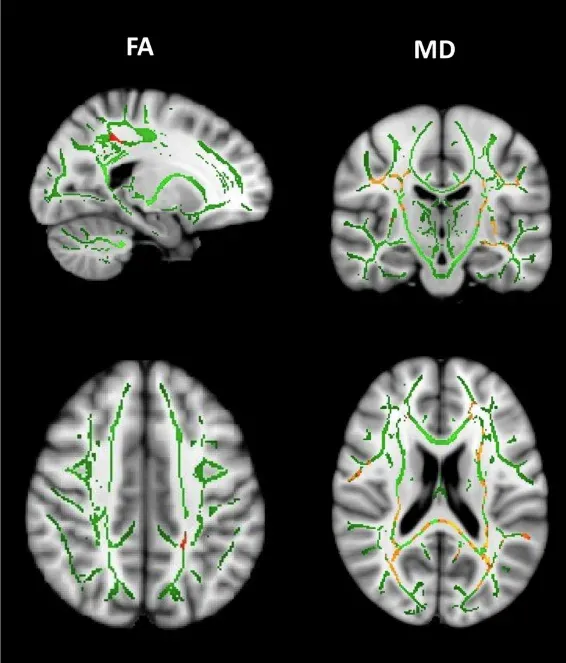
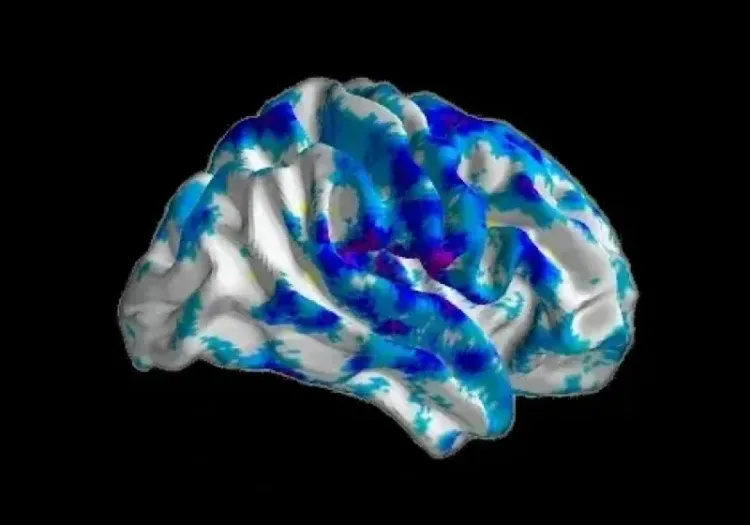
Longitudinal decreases in fractional anisotropy (FA) and increases in mean diffusivity (MD) between premanifest HD-gene mutation carriers and healthy controls. Over time, premanifest HD-gene mutation carriers show significant decreases in FA in the left superior longitudinal fasciculus. Additionally, widespread MD increases are observed in areas including the superior longitudinal fascicles, splenium of the corpus callosum, corona radiata, and external capsules. Figure reproduced from Estevez-Fraga et al. (Estevez-Fraga, 2023) under the Creative Commons Attribution License.
Overall, structural and diffusion MRI biomarkers offer significant insights into the progression of HD, with early indicators such as striatal atrophy and increased diffusivity in specific white matter tracts enabling early detection. These biomarkers not only aid in monitoring disease progression, but also provide essential tools for assessing the effectiveness of emerging therapies in clinical trials. By enabling stratification of subgroups according to disease stage, these biomarkers are critical for the evaluation of treatment effects at different stages of the disease (Klöppel, 2009). The observed changes in brain structure and microstructure reflect the complex and progressive nature of HD, underlining the need for reliable biomarkers to facilitate early diagnosis, intervention, and the assessment of therapeutic outcomes.
How do MRI biomarkers correlate with clinical outcomes in HD?
Regional brain volumes, particularly in the caudate and putamen, strongly correlate with subtle motor and cognitive impairments in individuals with expanded CAG repeats, even before a formal HD diagnosis (Kinnunen, 2021). This observation is true despite the reduced sensitivity of clinical scales in detecting early impairments in this population. Studies tracking biomarker changes have shown that subcortical atrophy, particularly reductions in caudate and putamen volumes, occurs prior to changes in clinical and cognitive biomarkers (Wijeratne, 2021).
In individuals diagnosed with HD, volumetric MRI and whole-brain voxel-based morphometry (VBM) results correlate with both motor and cognitive function, particularly in the caudate (Kinnunen, 2021). These correlations likely reflect the specialized functions of these brain regions in motor control and cognition. Additionally, striatal volumes, including those of the caudate and putamen, provide additional predictive information for disease progression, beyond traditional markers like age and CAG repeat length (Kinnunen, 2021).
Longitudinal studies indicate that changes in caudate and putamen volumes are often more prominent than clinical changes observed in motor or cognitive assessments. In a study examining the best predictors of disease progression, caudate and putamen volumes were identified as the most reliable indicators for tracking changes across different progression groups, as defined by the CAP score (Abeyasinghe, 2021). Notably, including additional motor, functional, and cognitive markers did not significantly improve the accuracy of predicting disease progression. These findings suggest that objective neuroimaging biomarkers like caudate and putamen volumes are more effective than subjective clinical assessments in distinguishing between disease stages (Abeyasinghe, 2021).
Additionally, studies have demonstrated that changes in diffusivity within corticostriatal pathways, particularly those connecting the putamen with the prefrontal cortex and primary motor cortex, are closely correlated with motor symptoms in HD-gene mutation carriers (Estevez-Fraga, 2020). Longitudinal changes in white matter integrity measured using diffusion tensor imaging (DTI) have also been shown to correlate with worsening motor symptoms measured by clinical scales, even in premanifest HD-gene mutation carriers (Estevez-Fraga, 2023). These findings highlight the significant role of early neuroimaging changes in predicting the future course of the disease.
In summary, neuroimaging biomarkers, particularly structural MRI and diffusion MRI, offer crucial insights into the early detection and monitoring of HD. Both striatal volumes and white matter integrity serve as valuable predictive markers for disease progression, even in the premanifest stages. These objective imaging measures are more reliable than traditional clinical scales in tracking early disease changes. Consequently, MRI biomarkers are critical tools for evaluating treatment outcomes in clinical trials, enabling more precise tracking of disease progression and therapeutic efficacy.
Our team would be happy to answer any questions about MRI Biomarkers in Clinical Trials of Huntington’s Disease or provide specific information about our other Imaging services.
Discover more about our Imaging Services
Related Content
Up-to-date information on best practices related to the use of neuroimaging in clinical trials of neurological diseases.
Spinocerebellar Ataxia Clinical Trials & Imaging Biomarkers
An overview of structural MRI, DTI and MRS imaging biomarkers to monitor disease progression in spinocerebellar ataxia clinical trials.
PET Imaging in Huntington’s Disease Clinical Trials
An overview of the use of PET imaging biomarkers for Huntington’s disease (HD) clinical trials.
Imaging Biomarkers for Friedreich’s Ataxia Clinical Trials
An overview of the use of MRI and DTI imaging biomarkers for Friedreich’s ataxia (FRDA) in research studies & multi-center clinical trials.
MRI in Clinical Trials of Multiple System Atrophy (MSA)
This resource provides an overview of the utility of volumetric MRI and diffusion-weighted imaging (DWI) as biomarkers in research studies of MSA.
Frontotemporal Dementia (FTD) & MRI Brain Atrophy
Neuroimaging biomarkers (including MRI brain atrophy) from the FTLDNI natural history study of Frontotemporal Dementia (FTD).
MRI & Corticobasal Degeneration (CBD)
Longitudinal MRI brain atrophy measures from the 4RTNI and FTLDNI studies including sample size calculations for clinical trials of Corticobasal Degeneration.