Biomarqueurs IRM dans les essais cliniques de la maladie de Huntington
Un aperçu de l'utilisation des biomarqueurs d'imagerie IRM structurelle et de diffusion pour les essais cliniques de la maladie de Huntington (MH).
Quelle est la présentation clinique et la base génétique de la MH?
Présentation clinique
La maladie de Huntington (MH) est un trouble neurodégénératif rare et progressif, hérité selon un mode autosomique dominant, se manifestant généralement par des symptômes d'apparition à l'âge adulte entre 35 et 40 ans. Son incidence est d'environ 4 à 10 pour 100 000 chez les individus d'origine européenne (Klöppel, 2009). Elle se caractérise principalement par la dégénérescence des neurones épineux moyens (MSNs) dans le striatum. Les études de neuroimagerie révèlent une atrophie précoce du noyau caudé et du putamen, ainsi qu'une perte progressive de la matière blanche cérébrale, détectable des années avant l'apparition des symptômes cliniques. À mesure que la maladie progresse, les individus subissent un déclin progressif du contrôle moteur, des fonctions cognitives et de la santé neuropsychiatrique.
Les symptômes moteurs de la MH impliquent des difficultés de mouvement involontaire et volontaire. Les mouvements involontaires incluent la chorée (mouvements irréguliers et saccadés), la dystonie (contractions musculaires) et la rigidité. Le contrôle moteur volontaire se détériore également, entraînant des symptômes tels que la dysarthrie (difficulté à parler), la dysphagie (difficulté à avaler) et l'akinesie (réduction de la capacité à initier le mouvement) (Jiang, 2023). Le déclin cognitif se caractérise par des déficits des fonctions exécutives, notamment des difficultés dans le comportement dirigé vers un but, ainsi que des altérations de l'apprentissage verbal, des capacités visuospatiales et, finalement, une perte de mémoire sévère. Les symptômes neuropsychiatriques sont également courants et peuvent inclure l'anxiété, l'irritabilité, la dépression, les comportements obsessionnels-compulsifs, l'agressivité, l'apathie et la psychose (Jiang, 2023). De plus, les individus atteints de MH souffrent souvent de perte de poids involontaire, de troubles du sommeil et de perturbations du rythme circadien.
L'apparition typique de la MH se situe entre 35 et 40 ans, bien qu'environ 5% des cas se présentent sous forme de maladie de Huntington juvénile (JHD), avec une apparition avant l'âge de 20 ans. La maladie est généralement décrite en trois stades. Le premier stade, connu sous le nom de stade présymptomatique (ou prémanifest), est marqué par l'absence de toute anomalie clinique notable. La phase suivante est la phase prodromique, où des changements subtils dans la fonction motrice, la cognition et le comportement commencent à apparaître. Le stade final est le stade manifeste, au cours duquel les symptômes deviennent prononcés et un diagnostic formel de MH est posé (Jiang, 2023).
Le diagnostic de la MH implique une combinaison de tests génétiques, d'évaluation des antécédents familiaux et d'évaluation clinique. L'échelle de notation de la maladie de Huntington unifiée (UHDRS) est fréquemment utilisée pour évaluer les symptômes moteurs, tandis que les méthodes de neuroimagerie, telles que l'imagerie par résonance magnétique (IRM), aident à exclure d'autres conditions et à surveiller la progression de la maladie. Une découverte clé en neuroimagerie est l'atrophie du noyau caudé, particulièrement sensible pour suivre la progression de la maladie et devenue un biomarqueur important dans les essais cliniques visant à évaluer de nouveaux traitements (Hobbs, 2024).
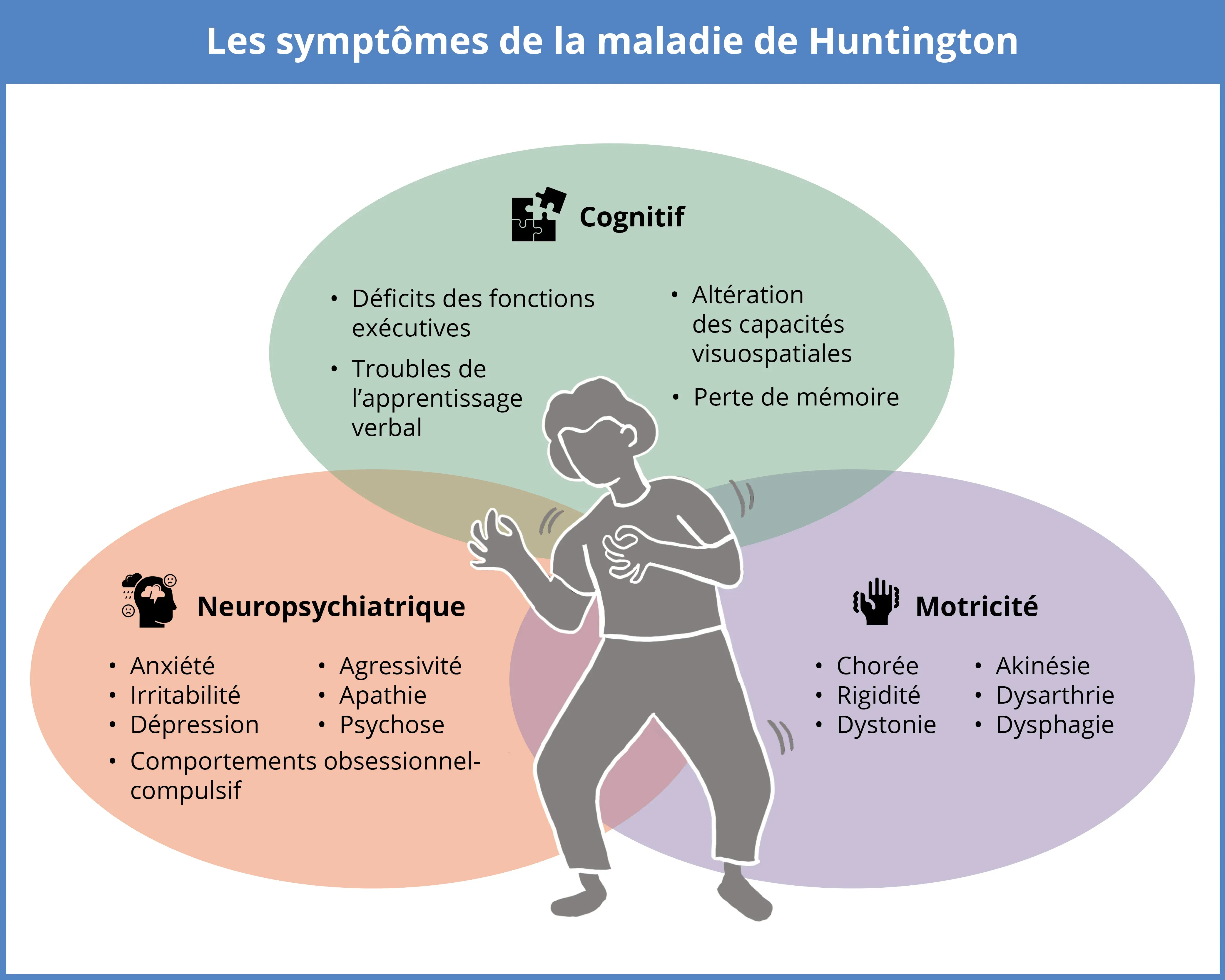
La maladie de Huntington (MH) est un trouble neurodégénératif rare et progressif caractérisé par une triade de symptômes, qui peuvent inclure un déclin cognitif (déficits des fonctions exécutives, altérations de l'apprentissage verbal et des compétences visuospatiales, et perte de mémoire), des changements neuropsychiatriques (anxiété, irritabilité, dépression, comportements obsessionnels-compulsifs, agressivité, apathie et psychose) et une dysfonction motrice (chorée, rigidité, dystonie, akinésie, dysarthrie et dysphagie).
 Cliquez pour copier le lien
Cliquez pour copier le lien
Base génétique
La MH résulte d'une mutation du gène HTT, qui code pour la protéine huntingtine. La mutation consiste en une expansion anormale de la répétition du trinucleotide CAG dans le premier exon du gène HTT sur le chromosome 4. Alors que le gène normal contient 10 à 35 répétitions, dans la MH, le nombre de répétitions dépasse un seuil critique, entraînant la production d'une forme toxique de la protéine huntingtine, appelée huntingtine mutante (mHTT) (Reiner, 2011). L'accumulation de mHTT perturbe les fonctions cellulaires, entraînant des dommages neuronaux. Les expansions plus grandes de la répétition CAG sont corrélées à une apparition plus précoce de la maladie et à une progression plus rapide, celles dépassant 39 répétitions étant considérées comme pleinement pénétrantes (Reiner, 2011; Wijeratne, 2021). considérées comme pleinement pénétrantes (Reiner, 2011 ; Wijeratne, 2021). La JHD, souvent associée à des expansions de répétition CAG supérieures à 60, se présente généralement avec des symptômes tels que la rigidité, les crises d'épilepsie et les changements comportementaux, contrairement à la MH à apparition adulte, où la chorée est plus courante (Reiner, 2011). La MH fait partie d'une classe plus large de troubles impliquant des expansions de répétition polyglutamine, y compris les ataxies spinocérébelleuses.
Le score produit CAG et âge (CAP) est un outil important utilisé pour évaluer la gravité de l'exposition à la mHTT et peut prédire la pathologie observée lors de l'autopsie. Ce système de notation aide à estimer la progression de la maladie et a montré une corrélation avec les résultats cliniques (Zhang, 2011).
Avec la découverte de la mutation du gène HTT, la MH est l'un des rares troubles neurodégénératifs pour lesquels un test génétique prédictif est disponible. Ce test permet aux individus ayant des antécédents familiaux de MH de déterminer s'ils portent la mutation génétique ou s'ils ont une MH présymptomatique (Andica, 2020). La capacité d'identifier les porteurs de la mutation du gène MH tôt offre des avantages significatifs, y compris la possibilité de participer à des essais cliniques axés sur les thérapies modifiant la maladie, y compris les traitements préventifs potentiels.
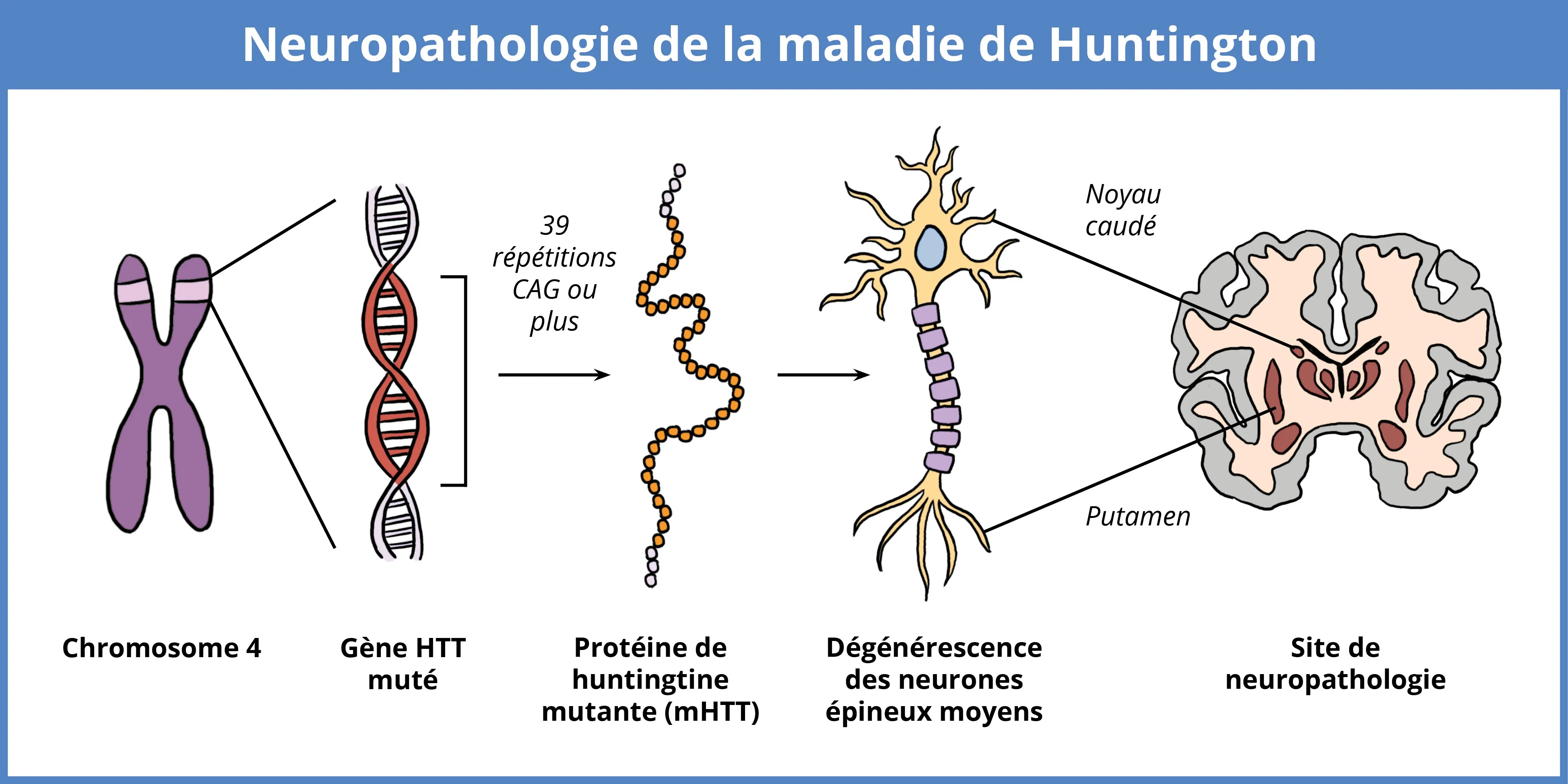
La MH est causée par une mutation du gène HTT situé sur le chromosome 4, caractérisée par une répétition CAG étendue. Alors que les individus normaux ont 10 à 35 répétitions, des expansions au-delà de 39 répétitions entraînent la production de la protéine huntingtine mutante toxique (mHTT). Les répétitions plus grandes sont liées à une apparition plus précoce de la maladie. L'accumulation de mHTT, en particulier dans les neurones épineux moyens (MSNs) du striatum, provoque une neurodégénérescence, entraînant des symptômes moteurs, cognitifs et neuropsychiatriques.
La manifestation clinique de la MH varie considérablement, avec des différences dans l'âge d'apparition, la présentation des symptômes, la progression de la maladie et la longueur de la répétition CAG contribuant à la complexité du trouble (Cao, 2024). Cette variabilité souligne la nécessité de biomarqueurs fiables pour permettre un diagnostic précoce et fournir des moyens précis et efficaces d'évaluer les résultats des traitements. Dans les essais cliniques, l'intégration des données de plusieurs biomarqueurs est cruciale pour stratifier de manière fiable les participants en sous-groupes et améliorer la précision des approches thérapeutiques (Wijeratne, 2018).
Quels biomarqueurs IRM sont efficaces pour suivre la progression de la maladie dans la MH?
Les changements neuropathologiques dans la maladie de Huntington (MH) sont les plus marqués dans le striatum, y compris le noyau caudé et le putamen. La caractéristique pathologique principale est la dégénérescence des neurones épineux moyens (MSNs) GABAergiques, qui sont essentiels pour le contrôle moteur et la fonction cognitive. À mesure que la MH progresse, la neurodégénérescence affecte également le cortex cérébral. Les études d'IRM structurelle et de diffusion ont montré que les anomalies cérébrales peuvent être détectées bien avant l'apparition des symptômes cliniques. L'IRM structurelle, par exemple, révèle des changements précoces comme l'atrophie striatale, tandis que l'IRM de diffusion identifie des altérations microstructurales dans des tractus spécifiques de la matière blanche (Reiner, 2011; Estevez-Fraga, 2023). Ces techniques d'imagerie jouent un rôle crucial dans la détection précoce de la MH et fournissent des informations importantes sur la progression de la maladie.
Biomarqueurs IRM structurelle
L'IRM volumétrique est un outil important pour surveiller la progression de la MH. L'atrophie striatale est considérée comme l'un des biomarqueurs les plus fiables et sensibles, car elle est fortement corrélée avec la longueur de la répétition CAG et l'âge auquel les symptômes cliniques se manifestent (Fazio, 2018; Kinnunen, 2021). Des études ont démontré que l'atrophie striatale peut être détectée jusqu'à 23 ans avant l'apparition des symptômes moteurs, ce qui en fait un biomarqueur précieux pour la surveillance de la maladie et les essais cliniques visant à évaluer les interventions thérapeutiques (Klöppel, 2009; Hobbs, 2024). L'atrophie précoce du noyau caudé et du putamen est associée à certains des plus grands effets parmi les biomarqueurs d'imagerie (Hobbs, 2015). Une étude examinant la progression de la maladie à l'aide de biomarqueurs de volume cérébral identifie le putamen et le noyau caudé comme les premières zones à présenter une atrophie (Wijeratne, 2018). À mesure que la MH progresse, l'atrophie se propage à d'autres régions du cerveau. L'amincissement cortical, en particulier dans des zones telles que les cortex occipital, moteur, préfrontal dorsomédial et pariétal, survient souvent avant l'apparition des symptômes moteurs (Kinnunen, 2021). De plus, le volume de la matière blanche cérébrale commence à diminuer tôt dans la maladie et continue de se détériorer à mesure que la maladie progresse (Kinnunen, 2021). En revanche, l'atrophie dans des régions comme le thalamus et l'hippocampe est moins prononcée (Fazio, 2018).
Biomarqueurs IRM de diffusion
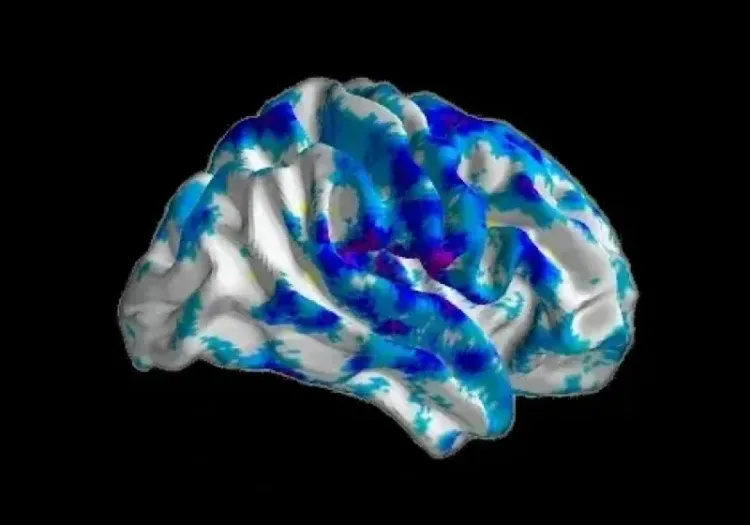
L'imagerie pondérée par diffusion (DWI) est particulièrement utile pour détecter les changements microstructuraux précoces dans la MH. Les études longitudinales montrent des réductions de l'anisotropie fractionnelle (FA) dans des tractus spécifiques de la matière blanche, tels que le faisceau longitudinal supérieur gauche dans la MH prémanifestée (Estevez-Fraga, 2023). De plus, une diminution de la FA dans le corps calleux peut être observée plusieurs années avant l'apparition des symptômes moteurs, soulignant le potentiel de l'IRM de diffusion en tant que biomarqueur précoce de la MH (Hobbs, 2012; Fazio, 2018). Ces réductions de la FA sont accompagnées d'augmentations de la diffusivité moyenne (MD) dans les faisceaux longitudinaux supérieurs, le splenium du corps calleux, la corona radiata et les capsules externes, suggérant un schéma spécifique de dégénérescence de la matière blanche dans la MH (Estevez-Fraga, 2023). Chez les porteurs de la mutation du gène MH prémanifestée, une augmentation de la MD est également observée dans les ganglions de la base et le thalamus (Fazio, 2018). À mesure que la maladie progresse vers son stade manifeste, les patients présentent une dégénérescence plus étendue, avec des réductions importantes de la FA et des augmentations de la MD observées dans les structures cérébrales profondes et superficielles (Estevez-Fraga, 2020). Les études longitudinales de diffusion montrent une augmentation de la MD, de la diffusivité axiale (AD) et de la diffusivité radiale (RD) dans le corps calleux, la corona radiata, le fornix, la matière blanche sous-corticale frontale et les capsules externes (Estevez-Fraga, 2023). Fait intéressant, certaines études ont rapporté des résultats incohérents dans les métriques de diffusion au sein des structures de la matière grise, ainsi que des augmentations inattendues de la FA et de la MD dans les régions striatales, des résultats qui nécessitent une enquête plus approfondie (Klöppel, 2009; Hobbs, 2012; Fazio, 2018; Hobbs, 2024).
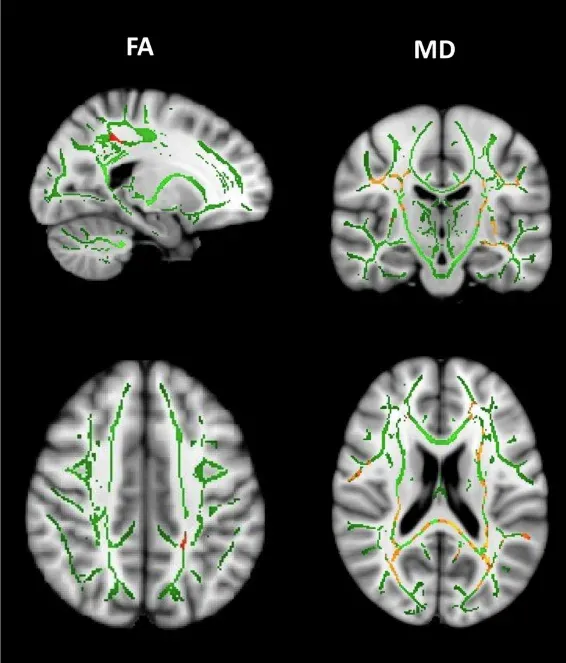
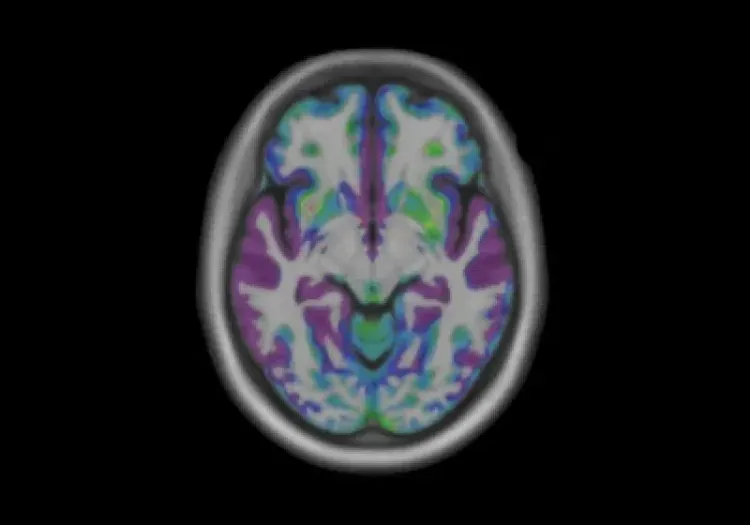
Diminutions longitudinales de l'anisotropie fractionnelle (FA) et augmentations de la diffusivité moyenne (MD) entre les porteurs de la mutation du gène MH prémanifestée et les témoins sains. Au fil du temps, les porteurs de la mutation du gène MH prémanifestée montrent des diminutions significatives de la FA dans le faisceau longitudinal supérieur gauche. De plus, des augmentations généralisées de la MD sont observées dans des zones incluant les faisceaux longitudinaux supérieurs, le splenium du corps calleux, la corona radiata et les capsules externes. Figure reproduite d'après Estevez-Fraga et al. (Estevez-Fraga, 2023) sous la licence Creative Commons Attribution License.
Dans l'ensemble, les biomarqueurs IRM structurelle et de diffusion offrent des informations significatives sur la progression de la MH, avec des indicateurs précoces tels que l'atrophie striatale et l'augmentation de la diffusivité dans des tractus spécifiques de la matière blanche permettant une détection précoce. Ces biomarqueurs non seulement aident à surveiller la progression de la maladie, mais fournissent également des outils essentiels pour évaluer l'efficacité des thérapies émergentes dans les essais cliniques. En permettant la stratification des sous-groupes en fonction du stade de la maladie, ces biomarqueurs sont essentiels pour l'évaluation des effets des traitements à différents stades de la maladie (Klöppel, 2009). Les changements observés dans la structure et la microstructure du cerveau reflètent la nature complexe et progressive de la MH, soulignant la nécessité de biomarqueurs fiables pour faciliter le diagnostic précoce, l'intervention et l'évaluation des résultats thérapeutiques.
Comment les biomarqueurs IRM corrèlent-ils avec les résultats cliniques dans la MH?
Les volumes cérébraux régionaux, en particulier dans le noyau caudé et le putamen, sont fortement corrélés avec des déficiences motrices et cognitives subtiles chez les individus avec des répétitions CAG étendues, même avant un diagnostic formel de la MH (Kinnunen, 2021). Cette observation est vraie malgré la sensibilité réduite des échelles cliniques pour détecter les déficiences précoces dans cette population. Les études suivant les changements des biomarqueurs ont montré que l'atrophie sous-corticale, en particulier les réductions des volumes du noyau caudé et du putamen, se produit avant les changements des biomarqueurs cliniques et cognitifs (Wijeratne, 2021).
Chez les individus diagnostiqués avec la MH, les résultats de l'IRM volumétrique et de la morphométrie par voxel de l'ensemble du cerveau (VBM) sont corrélés avec les fonctions motrices et cognitives, en particulier dans le noyau caudé (Kinnunen, 2021). Ces corrélations reflètent probablement les fonctions spécialisées de ces régions cérébrales dans le contrôle moteur et la cognition. De plus, les volumes striataux, y compris ceux du noyau caudé et du putamen, fournissent des informations prédictives supplémentaires pour la progression de la maladie, au-delà des marqueurs traditionnels tels que l'âge et la longueur de la répétition CAG (Kinnunen, 2021).
Les études longitudinales indiquent que les changements dans les volumes du noyau caudé et du putamen sont souvent plus prononcés que les changements cliniques observés dans les évaluations motrices ou cognitives. Dans une étude examinant les meilleurs prédicteurs de la progression de la maladie, les volumes du noyau caudé et du putamen ont été identifiés comme les indicateurs les plus fiables pour suivre les changements à travers différents groupes de progression, tels que définis par le score CAP (Abeyasinghe, 2021). Notamment, l'inclusion de marqueurs moteurs, fonctionnels et cognitifs supplémentaires n'a pas significativement amélioré la précision de la prédiction de la progression de la maladie. Ces résultats suggèrent que les biomarqueurs de neuroimagerie objectifs comme les volumes du noyau caudé et du putamen sont plus efficaces que les évaluations cliniques subjectives pour distinguer les stades de la maladie (Abeyasinghe, 2021).
De plus, des études ont démontré que les changements de diffusivité au sein des voies corticostriatales, en particulier celles reliant le putamen au cortex préfrontal et au cortex moteur primaire, sont étroitement corrélés avec les symptômes moteurs chez les porteurs de la mutation du gène MH (Estevez-Fraga, 2020). Les changements longitudinaux de l'intégrité de la matière blanche mesurés par l'imagerie par tenseur de diffusion (DTI) ont également montré une corrélation avec l'aggravation des symptômes moteurs mesurés par des échelles cliniques, même chez les porteurs de la mutation du gène MH prémanifestée (Estevez-Fraga, 2023). Ces résultats soulignent le rôle significatif des changements précoces de neuroimagerie dans la prédiction de l'évolution future de la maladie.
En résumé, les biomarqueurs de neuroimagerie, en particulier l'IRM structurelle et l'IRM de diffusion, offrent des informations cruciales pour la détection précoce et la surveillance de la MH. Les volumes striataux et l'intégrité de la matière blanche servent de marqueurs prédictifs précieux pour la progression de la maladie, même aux stades prémanifestés. Ces mesures d'imagerie objectives sont plus fiables que les échelles cliniques traditionnelles pour suivre les changements précoces de la maladie. Par conséquent, les biomarqueurs IRM sont des outils essentiels pour évaluer les résultats des traitements dans les essais cliniques, permettant un suivi plus précis de la progression de la maladie et de l'efficacité thérapeutique.
Notre équipe se fera un plaisir de répondre à vos questions sur les biomarqueurs IRM dans les essais cliniques de la maladie de Huntington ou de vous fournir des informations spécifiques sur nos autres services d'imagerie.
Découvrez nos services d'imagerie
Contenu connexe
Informations actualisées sur les meilleures pratiques liées à l'utilisation de la neuroimagerie dans les essais cliniques sur les maladies neurologiques.
Essais cliniques et biomarqueurs d'imagerie pour l'ataxie spinocérébelleuse
Vue d'ensemble des biomarqueurs d'imagerie IRM structurelle, DTI et MRS pour surveiller la progression de la maladie dans les essais cliniques sur l'ataxie spinocérébelleuse.
Biomarqueurs TEP dans les essais cliniques sur la maladie de Huntington
Un aperçu de l'utilisation des biomarqueurs d'imagerie TEP pour les essais cliniques de la maladie de Huntington (MH).
Biomarqueurs d'imagerie pour les essais cliniques sur l'ataxie de Friedreich
Un aperçu de l'utilisation des biomarqueurs d'imagerie IRM et DTI pour l'ataxie de Friedreich (FRDA) dans les études de recherche et les essais cliniques multicentriques.
L'IRM dans les essais cliniques sur l'atrophie du système multiple (MSA)
Cette ressource donne un aperçu de l'utilité de l'IRM volumétrique et de l'imagerie pondérée en diffusion (DWI) en tant que biomarqueurs dans les études de recherche sur la MSA.
Démence frontotemporale (DFT) et atrophie cérébrale par IRM
Biomarqueurs IRM (y compris l'atrophie cérébrale) issus des études FTLDNI sur l'histoire naturelle de la démence frontotemporale (DFT).
IRM et dégénérescence corticobasale (CBD)
Mesures longitudinales de l'atrophie cérébrale par IRM provenant des études 4RTNI et FTLDNI, y compris le calcul de la taille des échantillons pour les essais cliniques sur la dégénérescence corticobasale.